
A Transcriptome Survey Spanning Life Stages and Sexes of the Harlequin Bug, Murgantia histrionica


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
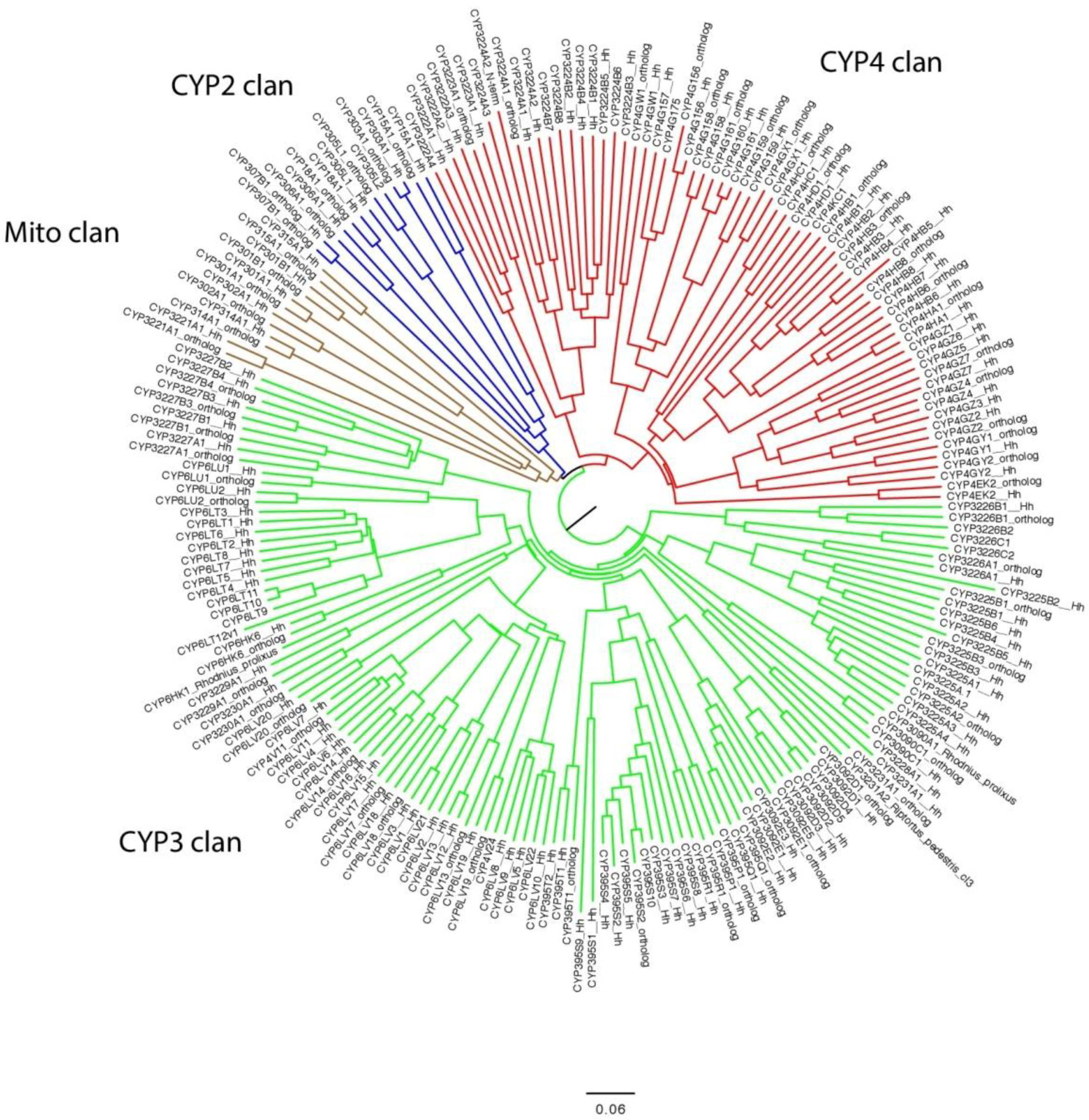
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aldrich, J.R.; Avery, J.W.; Lee, C.J.; Graf, J.C.; Harrison, D.J.; Bin, F. Semiochemistry of cabbage bugs (Heteroptera: Pentatomidae: Eurydema and Murgantia). J. Entomol. Sci. 1996, 31, 172–182. [Google Scholar]
- Chittenden, F.H. Harlequin Cabbage Bug and Its Control; USDA Farmers’ Bulletins (U.S. Department of Agriculture): Washington, DC, USA, 1920.
- Wallingford, A.K.; Kuhar, T.P.; Schultz, P.B.; Freeman, J.H. Harlequin bug biology and pest management in brassicaceous crops. J. Integr. Pest Manag. 2011, 2, H1–H4. [Google Scholar] [CrossRef]
- Walker, H.G.; Anderson, L.D. Report on the control of the harlequin bug, Murgantia histrionica Hahn, with notes on the severity of an outbreak of this insect in 1932. J. Econ. Entomol. 1933, 26, 129–135. [Google Scholar] [CrossRef]
- DiMeglio, A.S.; Wallingford, A.K.; Weber, D.C.; Kuhar, T.P.; Mullins, D. Supercooling points of Murgantia histrionica (Hemiptera: Pentatomidae) and field mortality in the mid-Atlantic United States following lethal low temperatures. Environ. Entomol. 2016, 45, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.L.; Pezzini, D.T.; Michel, A.P.; Hunt, T.E. Identification, Biology, Impacts, and Management of Stink Bugs (Hemiptera: Heteroptera: Pentatomidae) of Soybean and Corn in the Midwestern United States. J. Integr. Pest Manag. 2017, 8, 11. [Google Scholar] [CrossRef]
- Lockwood, J.A. Six-Legged Soldiers: Using Insects as Weapons of War; Oxford University Press: Oxford, NY, USA, 2010. [Google Scholar]
- Capinera, J.L. Harlequin Bug, Murgantia histrionica (Hahn) (Hemiptera: Pentatomidae). In Encyclopedia of Entomology; Capinera, J.L., Ed.; Springer: Dordrecht, The Netherlands, 2008; pp. 1766–1768. [Google Scholar]
- Aliabadi, A.; Renwick, J.A.A.; Whitman, D.W. Sequestration of glucosinolates by harlequin bug Murgantia histrionica. J. Chem. Ecol. 2002, 28, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Joga, M.R.; Zotti, M.J.; Smagghe, G.; Christiaens, O. RNAi efficiency, systemic properties, and novel delivery methods for pest insect control: What we know so far. Front. Physiol. 2016, 7, 553. [Google Scholar] [CrossRef] [PubMed]
- Zahn, D.K.; Moreira, J.A.; Millar, J.G. Identification, synthesis, and bioassay of a male-specific aggregation pheromone from the harlequin bug, Murgantia histrionica. J. Chem. Ecol. 2008, 34, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Zahn, D.K.; Moreira, J.A.; Millar, J.G. Erratum to: Identification, synthesis, and bioassay of a male-specific aggregation pheromone from the harlequin bug, Murgantia histrionica. J. Chem. Ecol. 2012, 38, 126. [Google Scholar] [CrossRef]
- Khrimian, A.; Shirali, S.; Vermillion, K.E.; Siegler, M.A.; Guzman, F.; Chauhan, K.; Aldrich, J.R.; Weber, D.C. Determination of the stereochemistry of the aggregation pheromone of harlequin bug, Murgantia histrionica. J. Chem. Ecol. 2014, 40, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.C.; Walsh, G.C.; DiMeglio, A.S.; Athanas, M.M.; Leskey, T.C.; Khrimian, A. Attractiveness of harlequin bug, Murgantia histrionica, aggregation pheromone: Field response to isomers, ratios, and dose. J. Chem. Ecol. 2014, 40, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G.C.; Dimeglio, A.S.; Khrimian, A.; Weber, D.C. Marking and retention of harlequin bug, Murgantia histrionica (Hahn) (Hemiptera: Pentatomidae), on pheromone-baited and unbaited plants. J. Pest Sci. 2016, 89, 21–29. [Google Scholar] [CrossRef]
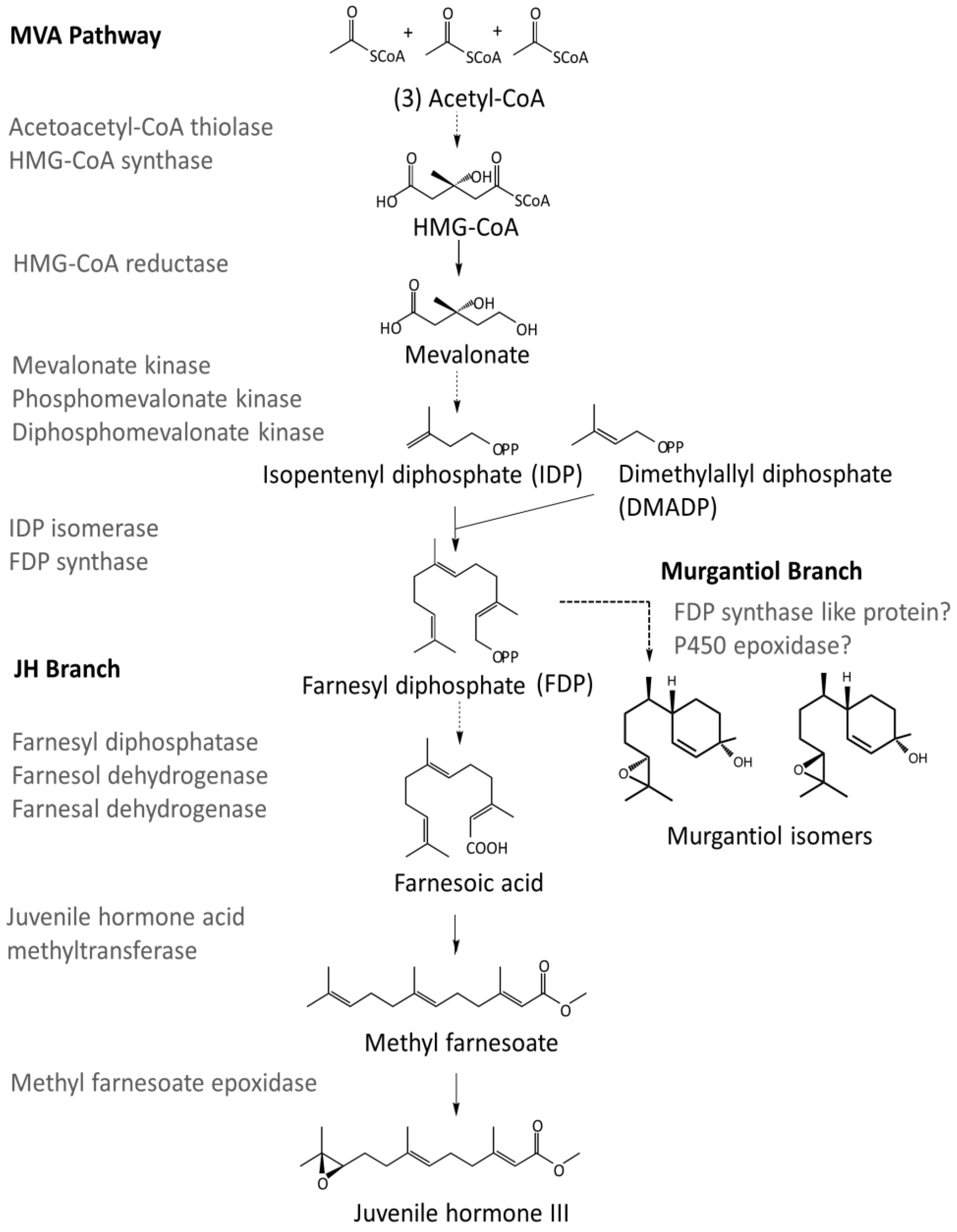
- Bellés, X.; Martín, D.; Piulachs, M.-D. The mevalonate pathway and the synthesis of juvenile hormone in insects. Annu. Rev. Entomol. 2005, 50, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Beran, F.; Rahfeld, P.; Luck, K.; Nagel, R.; Vogel, H.; Wielsch, N.; Irmisch, S.; Ramasamy, S.; Gershenzon, J.; Heckel, D.G.; et al. Novel family of terpene synthases evolved from trans-isoprenyl diphosphate synthases in a flea beetle. Proc. Natl. Acad. Sci. USA 2016, 113, 2922–2927. [Google Scholar] [CrossRef] [PubMed]
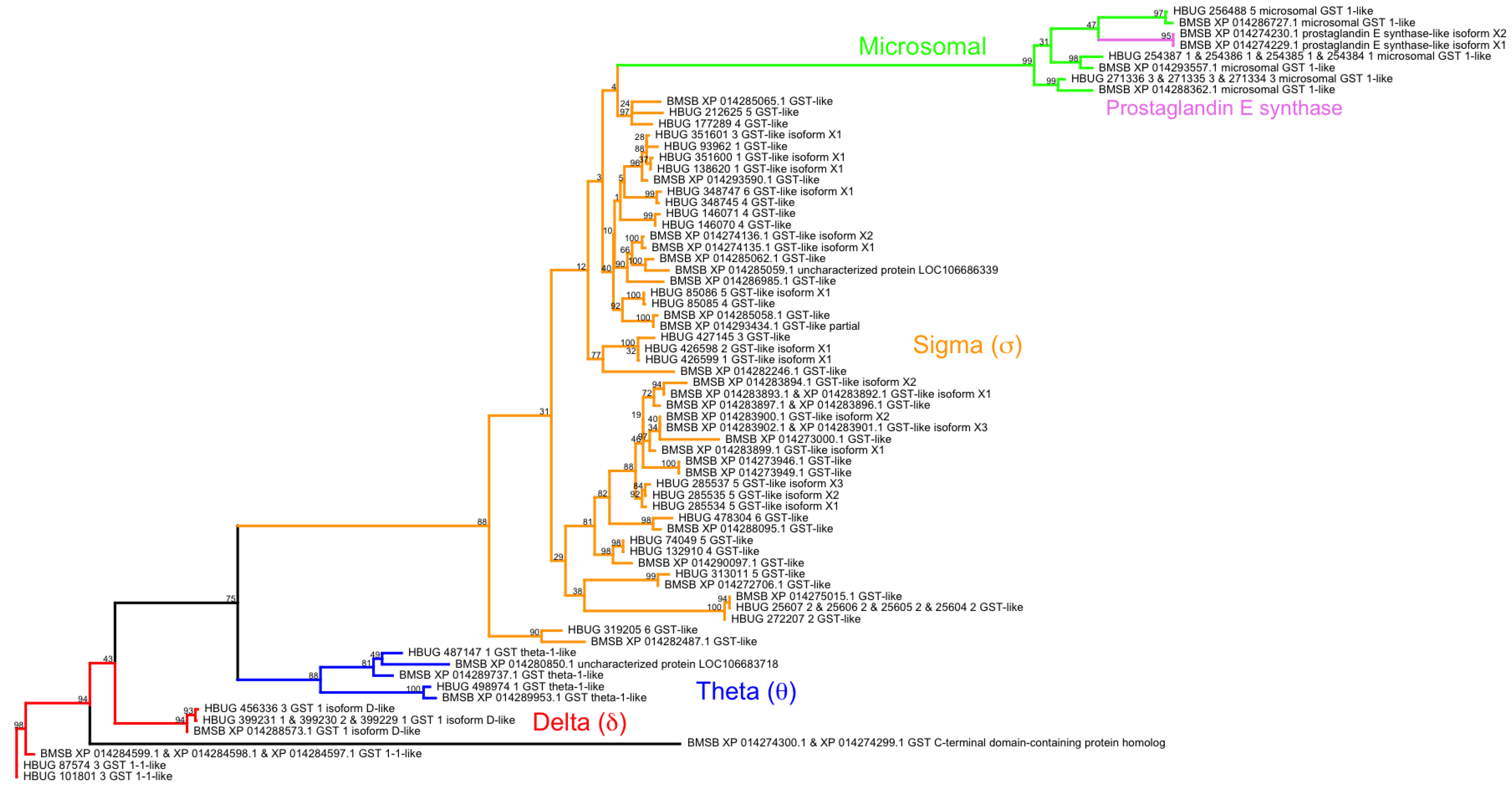
- Friedman, R. Genomic organization of the glutathione S-transferase family in insects. Mol. Phylogenet. Evol. 2011, 61, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Moores, G.D.; Devine, G.J.; Devonshire, A.L. Insecticide-insensitive acetylcholinesterase can enhance esterase-based resistance in Myzus persicae and Myzus nicotianae. Pestic. Biochem. Physiol. 1994, 49, 114–120. [Google Scholar] [CrossRef]
- Moores, G.D.; Gao, X.; Denholm, I.; Devonshire, A.L. Characterisation of insensitive acetylcholinesterase in insecticide-resistant cotton aphids, Aphis gossypii Glover (Homoptera: Aphididae). Pestic. Biochem. Physiol. 1996, 56, 102–110. [Google Scholar] [CrossRef]
- Oakeshott, J.G.; Johnson, R.M.; Berenbaum, M.R.; Ranson, H.; Cristino, A.S.; Claudianos, C. Metabolic enzymes associated with xenobiotic and chemosensory responses in Nasonia vitripennis. Insect Mol. Biol. 2010, 19 (Suppl. 1), 147–163. [Google Scholar] [CrossRef] [PubMed]
- Claudianos, C.; Ranson, H.; Johnson, R.M.; Biswas, S.; Schuler, M.A.; Berenbaum, M.R.; Feyereisen, R.; Oakeshott, J.G. A deficit of detoxification enzymes: Pesticide sensitivity and environmental response in the honeybee. Insect Mol. Biol. 2006, 15, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Oakeshott, J.G.; Devonshire, A.L.; Claudianos, C.; Sutherland, T.D.; Horne, I.; Campbell, P.M.; Ollis, D.L.; Russell, R.J. Comparing the organophosphorus and carbamate insecticide resistance mutations in cholin- and carboxyl-esterases. Chem. Biol. Interact. 2005, 157–158, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Rewitz, K.F.; O’Connor, M.B.; Gilbert, L.I. Molecular evolution of the insect Halloween family of cytochrome P450s: Phylogeny, gene organization and functional conservation. Insect Biochem. Mol. Biol. 2007, 37, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Kenny, N.J.; Lam, H.M.; Chan, T.F.; Chu, K.H.; Bendena, W.G.; Tobe, S.S.; Hui, J.H.L. How did arthropod sesquiterpenoids and ecdysteroids arise? Comparison of hormonal pathway genes in noninsect arthropod genomes. Genome Biol. Evol. 2015, 7, 1951–1959. [Google Scholar] [PubMed]
- Qiu, Y.; Tittiger, C.; Wicker-Thomas, C.; Le Goff, G.; Young, S.; Wajnberg, E.; Fricaux, T.; Taquet, N.; Blomquist, G.J.; Feyereisen, R. An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 14858–14863. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, X.; Wang, Y.; Moussian, B.; Zhu, K.Y.; Li, S.; Ma, E.; Zhang, J. LmCYP4G102: An oenocyte-specific cytochrome P450 gene required for cuticular waterproofing in the migratory locust, Locusta migratoria. Sci. Rep. 2016, 6, 29980. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, M.; Gong, Y.; Liu, F.; Li, T. Cytochrome P450s—Their expression, regulation, and role in insecticide resistance. Pestic. Biochem. Physiol. 2015, 120, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Edi, C.V.; Djogbénou, L.; Jenkins, A.M.; Regna, K.; Muskavitch, M.A.T.; Poupardin, R.; Jones, C.M.; Essandoh, J.; Kétoh, G.K.; Paine, M.J.I.; et al. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS Genet. 2014, 10, e1004236. [Google Scholar] [CrossRef] [PubMed]
- David, J.-P.; Ismail, H.M.; Chandor-Proust, A.; Paine, M.J.I. Role of cytochrome P450s in insecticide resistance: Impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120429. [Google Scholar] [CrossRef] [PubMed]
- Balabanidou, V.; Kampouraki, A.; MacLean, M.; Blomquist, G.J.; Tittiger, C.; Juárez, M.P.; Mijailovsky, S.J.; Chalepakis, G.; Anthousi, A.; Lynd, A.; et al. Cytochrome P450 associated with insecticide resistance catalyzes cuticular hydrocarbon production in Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2016, 113, 9268–9273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Widemann, E.; Bernard, G.; Lesot, A.; Pinot, F.; Pedrini, N.; Keyhani, N.O. CYP52X1, representing new cytochrome P450 subfamily, displays fatty acid hydroxylase activity and contributes to virulence and growth on insect cuticular substrates in entomopathogenic fungus Beauveria bassiana. J. Biol. Chem. 2012, 287, 13477–13486. [Google Scholar] [CrossRef] [PubMed]
- Maïbèche-Coisne, M.; Nikonov, A.A.; Ishida, Y.; Jacquin-Joly, E.; Leal, W.S. Pheromone anosmia in a scarab beetle induced by in vivo inhibition of a pheromone-degrading enzyme. Proc. Natl. Acad. Sci. USA 2004, 101, 11459–11464. [Google Scholar] [CrossRef] [PubMed]
- Blomquist, G.J.; Figueroa-Teran, R.; Aw, M.; Song, M.; Gorzalski, A.; Abbott, N.L.; Chang, E.; Tittiger, C. Pheromone production in bark beetles. Insect Biochem. Mol. Biol. 2010, 40, 699–712. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhang, Y.-F.; Hong, D.-Y.; Wang, J.; Wang, X.-L.; Zuo, L.-H.; Tang, X.-F.; Xu, W.-M.; He, M. A reference gene set for sex pheromone biosynthesis and degradation genes from the diamondback moth, Plutella xylostella, based on genome and transcriptome digital gene expression analyses. BMC Genom. 2017, 18, 219. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Gowin, J.; Hartfelder, K.; Korb, J. The scent of royalty: A P450 gene signals reproductive status in a social insect. Mol. Biol. Evol. 2014, 31, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Vakatov, D. The NCBI C++ Toolkit Book [Internet]; National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sparks, M.E.; Blackburn, M.B.; Kuhar, D.; Gundersen-Rindal, D.E. Transcriptome of the Lymantria dispar (Gypsy Moth) Larval Midgut in Response to Infection by Bacillus thuringiensis. PLoS ONE 2013, 8, e61190. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.E.; Shelby, K.S.; Kuhar, D.; Gundersen-Rindal, D.E. Transcriptome of the invasive brown marmorated stink bug, Halyomorpha halys (Stål) (Heteroptera: Pentatomidae). PLoS ONE 2014, 9, e111646. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Gremme, G.; Brendel, V.; Sparks, M.E.; Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 2005, 47, 965–978. [Google Scholar] [CrossRef]
- Sparks, M.E.; Gundersen-Rindal, D.E.; Harrison, R.L. Complete Genome Sequence of a Novel Iflavirus from the Transcriptome of Halyomorpha halys, the Brown Marmorated Stink Bug. Genome Announc. 2013, 1, e00910-13. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, L.J.; Meulia, T.; Sabree, Z.L. Habitat visualization and genomic analysis of “Candidatus Pantoea carbekii,” the primary symbiont of the brown marmorated stink bug. Genome Biol. Evol. 2015, 7, 620–635. [Google Scholar] [CrossRef] [PubMed]
- Prado, S.S.; Almeida, R.P.P. Phylogenetic placement of pentatomid stink bug gut symbionts. Curr. Microbiol. 2009, 58, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Duron, O.; Noël, V. A wide diversity of Pantoea lineages are engaged in mutualistic symbiosis and cospeciation processes with stinkbugs. Environ. Microbiol. Rep. 2016, 8, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Panizzi, A.R.; McPherson, J.E.; James, D.G.; Javahery, M.; McPherson, R.M. Economic importance of stink bugs (Pentatomidae). In Heteroptera of Economic Importance; Schaefer, C.W., Panizzi, A.R., Eds.; CRC Press: Boca Raton, FL, USA, 2000; pp. 421–474. [Google Scholar]
- McPherson, J.E. Invasive Stink Bugs and Related Species (Pentatomoidea): Biology, Higher Systematics, Semiochemistry, and Management; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- De Clercq, P. Predaceous stinkbugs (Pentatomidae: Asopinae). In Heteroptera of Economic Importance; Schaefer, C.W., Panizzi, A.R., Eds.; CRC Press: Boca Raton, FL, USA, 2000; pp. 737–789. [Google Scholar]
- Ramsey, J.S.; Rider, D.S.; Walsh, T.K.; De Vos, M.; Gordon, K.H. J.; Ponnala, L.; Macmil, S.L.; Roe, B.A.; Jander, G. Comparative analysis of detoxification enzymes in Acyrthosiphon pisum and Myzus persicae. Insect Mol. Biol. 2010, 19 (Suppl. 2), 155–164. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.A.; Subramanian, G.M.; Halpern, A.; Sutton, G.G.; Charlab, R.; Nusskern, D.R.; Wincker, P.; Clark, A.G.; Ribeiro, J.M.C.; Wides, R.; et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 2002, 298, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Ranson, H.; Claudianos, C.; Ortelli, F.; Abgrall, C.; Hemingway, J.; Sharakhova, M.V.; Unger, M.F.; Collins, F.H.; Feyereisen, R. Evolution of supergene families associated with insecticide resistance. Science 2002, 298, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Devonshire, A.L.; Foster, G.N.; Sawicki, R.M. Peach-potato aphid, Myzus persicae (Sulz.), resistant to organophosphorus and carbamate insecticides on potatoes in Scotland. Plant Pathol. 1977, 26, 60–62. [Google Scholar] [CrossRef]
- Devonshire, A.L.; Sawicki, R.M. Insecticide-resistant Myzus persicae as an example of evolution by gene duplication. Nature 1979, 280, 140–141. [Google Scholar] [CrossRef]
- Noriega, F.G.; Ribeiro, J.M.C.; Koener, J.F.; Valenzuela, J.G.; Hernandez-Martinez, S.; Pham, V.M.; Feyereisen, R. Comparative genomics of insect juvenile hormone biosynthesis. Insect Biochem. Mol. Biol. 2006, 36, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Kinjoh, T.; Kaneko, Y.; Itoyama, K.; Mita, K.; Hiruma, K.; Shinoda, T. Control of juvenile hormone biosynthesis in Bombyx mori: Cloning of the enzymes in the mevalonate pathway and assessment of their developmental expression in the corpora allata. Insect Biochem. Mol. Biol. 2007, 37, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Nouzova, M.; Edwards, M.J.; Mayoral, J.G.; Noriega, F.G. A coordinated expression of biosynthetic enzymes controls the flux of juvenile hormone precursors in the corpora allata of mosquitoes. Insect Biochem. Mol. Biol. 2011, 41, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Noriega, F.G. Juvenile hormone biosynthesis in insects: What is new, what do we know, and what questions remain? Int. Sch. Res. Notices. 2014. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Sequence Data | Data Post-Normalization | |||
|---|---|---|---|---|
| read pairs | bases | read pairs | bases | |
| 2nd Instar | 213,817,714 | 42,763,542,800 | 13,268,461 | 2,653,692,200 |
| 4th Instar | 222,117,185 | 44,423,437,000 | 15,199,439 | 3,039,887,800 |
| Female | 205,127,388 | 41,025,477,600 | 11,786,560 | 2,357,312,000 |
| Male | 218,215,787 | 43,643,157,400 | 14,191,526 | 2,838,305,200 |
| Totals | 859,278,074 | 171,855,614,800 | 54,445,986 | 10,889,197,200 |
| Gold-Tier PUTs (Trimmed per Homology Info) | All PUT Sequences (Untrimmed) | ||||
|---|---|---|---|---|---|
| Pfam hit | Pfam Description | Counts | Pfam hit | Pfam Description | Counts |
| PF00069.23 | Protein kinase domain | 511 | PF00078.25 | Reverse transcriptase (RNA-dependent DNA polymerase) | 3272 |
| PF07714.15 | Protein tyrosine kinase | 504 | PF00096.24 | Zinc finger, C2H2 type | 1424 |
| PF13894.4 | C2H2-type zinc finger | 383 | PF13894.4 | C2H2-type zinc finger | 1292 |
| PF00096.24 | Zinc finger, C2H2 type | 381 | PF13465.4 | Zinc-finger double domain | 1262 |
| PF13857.4 | Ankyrin repeats (many copies) | 324 | PF00069.23 | Protein kinase domain | 1256 |
| PF13465.4 | Zinc-finger double domain | 323 | PF12796.5 | Ankyrin repeats (3 copies) | 1245 |
| PF12796.5 | Ankyrin repeats (3 copies) | 323 | PF13637.4 | Ankyrin repeats (many copies) | 1209 |
| PF00400.30 | WD domain, G-beta repeat | 323 | PF13857.4 | Ankyrin repeats (many copies) | 1203 |
| PF13637.4 | Ankyrin repeats (many copies) | 319 | PF07714.15 | Protein tyrosine kinase | 1199 |
| PF00023.28 | Ankyrin repeat | 316 | PF07690.14 | Major Facilitator Superfamily | 1183 |
| PF13606.4 | Ankyrin repeat | 313 | PF00023.28 | Ankyrin repeat | 1171 |
| PF07690.14 | Major Facilitator Superfamily | 288 | PF13606.4 | Ankyrin repeat | 1154 |
| PF12894.5 | Anaphase-promoting complex subunit 4 WD40 domain | 280 | PF13909.4 | C2H2-type zinc-finger domain | 956 |
| PF13927.4 | Immunoglobulin domain | 268 | PF01359.16 | Transposase (partial DDE domain) | 844 |
| PF14531.4 | Kinase-like | 266 | PF00400.30 | WD domain, G-beta repeat | 834 |
| PF13895.4 | Immunoglobulin domain | 261 | PF13927.4 | Immunoglobulin domain | 741 |
| PF00047.23 | Immunoglobulin domain | 259 | PF00083.22 | Sugar (and other) transporter | 734 |
| PF07679.14 | Immunoglobulin I-set domain | 257 | PF07679.14 | Immunoglobulin I-set domain | 676 |
| PF01926.21 | 50S ribosome-binding GTPase | 256 | PF00047.23 | Immunoglobulin domain | 636 |
| PF07686.15 | Immunoglobulin V-set domain | 255 | PF13895.4 | Immunoglobulin domain | 635 |
| PF08477.11 | Ras of Complex, Roc, domain of DAPkinase | 244 | PF00005.25 | ABC transporter | 620 |
| PF00071.20 | Ras family | 238 | PF00076.20 | RNA recognition motif (a.k.a. RRM, RBD, or RNP domain) | 611 |
| PF00076.20 | RNA recognition motif (a.k.a. RRM, RBD, or RNP domain) | 235 | PF00067.20 | Cytochrome P450 | 590 |
| PF12799.5 | Leucine Rich repeats (2 copies) | 234 | PF00665.24 | Integrase core domain | 578 |
| PF00025.19 | ADP-ribosylation factor family | 211 | PF07686.15 | Immunoglobulin V-set domain | 546 |
| 2nd Instar Expression Relative to 4th Instar | ||||
| log2FC | Direction | 2nd Instar | 4th Instar | NR Gene |
| 5.1284 | up | 253.24 | 7.24 | adult-specific cuticular protein ACP-20-like |
| 4.6366 | up | 1055.89 | 42.45 | uncharacterized protein LOC106688964 |
| 3.6726 | up | 366.36 | 28.73 | tubulin beta-1 chain |
| 3.5047 | up | 380.01 | 33.48 | protein takeout-like |
| 3.3727 | up | 192.25 | 18.56 | GTP cyclohydrolase 1 isoform X1 |
| 5.3563 | down | 8.17 | 334.68 | heat shock 70 kDa protein cognate 4 |
| 5.3279 | down | 7.17 | 287.99 | acyl-CoA Delta(11) desaturase-like isoform X1 |
| 5.2056 | down | 6.33 | 233.59 | uncharacterized protein LOC106679388 |
| 4.7751 | down | 9.21 | 252.18 | lysosomal alpha-mannosidase |
| 4.6475 | down | 6.05 | 151.63 | uncharacterized protein LOC106683993 |
| Female Expression Relative to Male | ||||
| log2FC | Direction | Female | Male | NR Gene |
| 4.1357 | up | 375.11 | 21.34 | heat shock 70 kDa protein cognate 4 |
| 3.5908 | up | 104.22 | 8.65 | JH acid O-methyltransferase-like isoform X1 |
| 2.8439 | up | 88.45 | 12.32 | uncharacterized protein LOC106679932 |
| 2.7760 | up | 124.18 | 18.13 | troponin C, isoform 1-like |
| 2.7262 | up | 34.21 | 5.17 | adult-specific cuticular protein ACP-20-like |
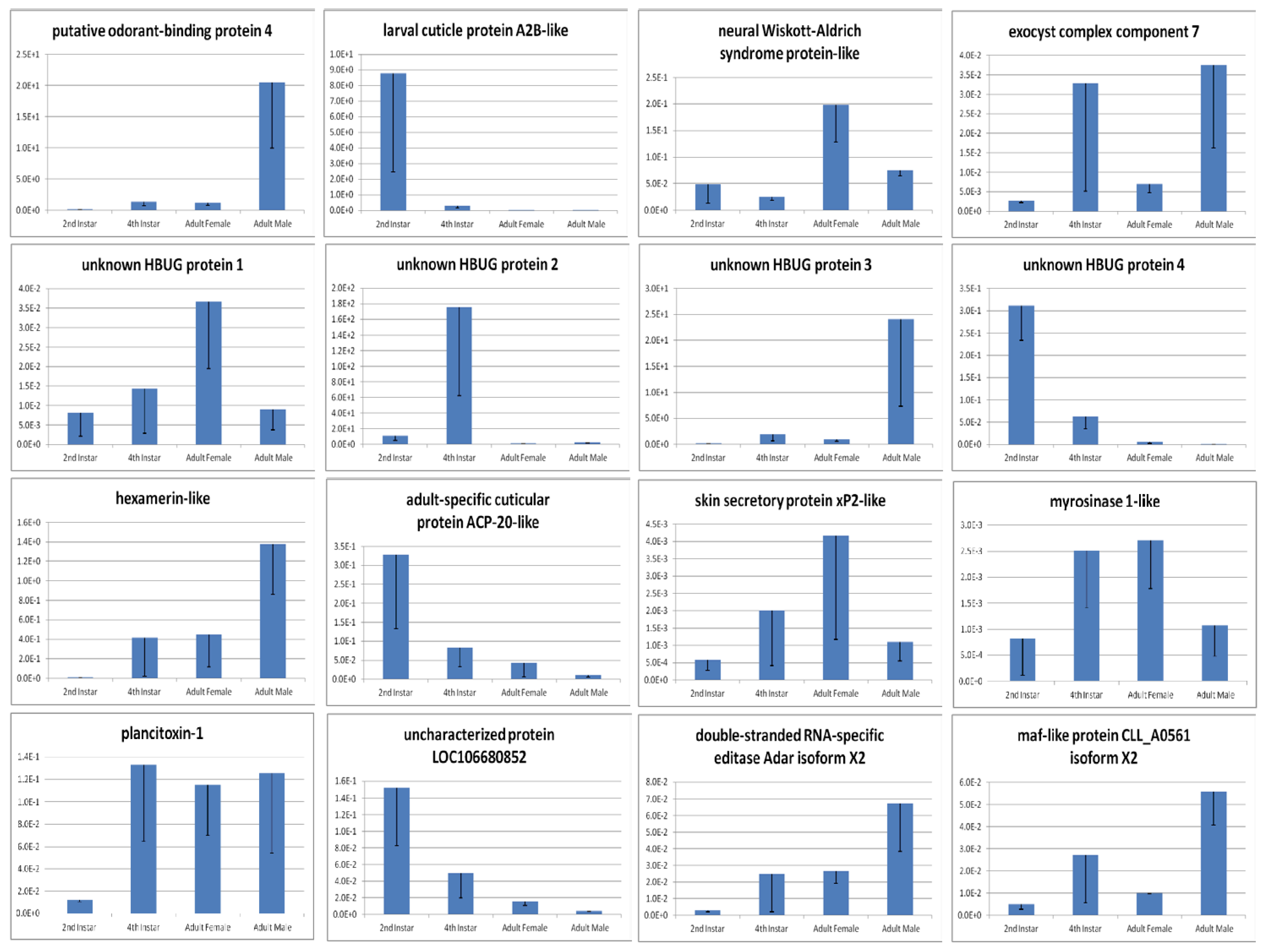
| 3.9320 | down | 570.39 | 8706.01 | putative odorant-binding protein 4 |
| 2.7080 | down | 5.58 | 36.46 | E3 ubiquitin-protein ligase TRIM37-like isoform X2 |
| 2.2543 | down | 6.29 | 30.01 | exocyst complex component 7 |
| 1.9412 | down | 6.51 | 25.00 | DDB1- and CUL4-associated factor 6-like |
| 1.9045 | down | 7.18 | 26.88 | exosome complex component CSL4 |
| Adult Expression Relative to Nymph | ||||
| log2FC | Direction | Adult | Nymph | NR Gene |
| 3.8701 | up | 5288.23 | 361.66 | putative odorant-binding protein 4 |
| 3.7040 | up | 105.04 | 8.06 | inositol monophosphatase 2-like |
| 3.4889 | up | 98.80 | 8.80 | uncharacterized protein LOC106686819 |
| 3.4091 | up | 78.93 | 7.43 | uncharacterized protein LOC106691169 |
| 3.0628 | up | 264.22 | 31.62 | NADP-dependent malic enzyme-like isoform X3 |
| 6.0853 | down | 44.80 | 3041.89 | uncharacterized protein LOC106688633 |
| 4.3486 | down | 5.41 | 110.22 | pro-resilin-like |
| 3.7376 | down | 13.83 | 184.48 | probable antibacterial peptide |
| 3.6876 | down | 9.00 | 115.96 | cuticle protein 18.6, isoform B |
| 3.2451 | down | 11.07 | 104.96 | GTP cyclohydrolase 1 isoform X1 |
| ♂ (TPM) | ♀ (TPM) | |log2(♂:♀)| | Direction | HBUG PUT Id | Best NR hit |
| 0.03 | 18.87 | 9.2969 | - | 472510 | XP_014270541.1 (CYP4GZ4) |
| 0.07 | 24.35 | 8.4424 | - | 473495 | XP_014286441.1 (CYP6LT5) |
| 0.40 | 72.44 | 7.5006 | - | 419699 | XP_014273208.1 (CYP6LT7) |
| 0.06 | 8.73 | 7.1849 | - | 103772 | XP_014286439.1 (CYP6LT3) |
| 0.27 | 19.68 | 6.1876 | - | 103769 | XP_014286439.1 (CYP6LT3) |
| 6.35 | 0.00 | undefined | + | 478727 | XP_014293876.1 (CYP307B1) |
| 4.71 | 0.00 | undefined | + | 173857 | XP_014285590.1 (CYP3226B1) |
| 9.95 | 0.01 | 9.9586 | + | 298833 | XP_014293876.1 (CYP307B1) |
| 4.30 | 0.07 | 5.9408 | + | 137898 | XP_014288222.1 (CYP3227B4) |
| 3.26 | 0.15 | 4.4418 | + | 521147 | XP_014276563.1 (CYP3225B3) |
| 5.85 | 0.61 | 3.2616 | + | 428429 | XP_014279285.1 (CYP302A1) |
| 38.09 | 6.24 | 2.6098 | + | 103771 | XP_014286439.1 (CYP6LT3) |
| 21.99 | 6.92 | 1.6680 | + | 504543 | XP_014284935.1 (CYP4HB7) |
| 15.69 | 5.20 | 1.5933 | + | 456532 | XP_014285589.1 (CYP3226B1) |
| 25.99 | 8.88 | 1.5493 | + | 486339 | XP_014285590.1 (CYP3226B1) |
| 14.45 | 5.10 | 1.5025 | + | 316472 | XP_014274999.1 (CYP3224A2) |
| 15.06 | 5.51 | 1.4506 | + | 165088 | XP_014271425.1 (CYP6LU1) |
| 13.48 | 6.26 | 1.1066 | + | 212119 | XP_014284933.1 (CYP4HB6) |
| 7.51 | 15.33 | 1.0295 | - | 504542 | XP_014284935.1 (CYP4HB7) |
| Enzyme Name | Query Sequences (multiple taxa) | Transcript Identified (M. histrionica) | Blastx Support (H. halys) |
|---|---|---|---|
| Acetoacetyl-CoA thiolase | XM_014419845,XM_014386017,XM_015512081,AK403218 | HBug_USDA-ARS_IIBBL.267134 | XP_014294739.1 |
| HBug_USDA-ARS_IIBBL.43170 | XP_014277769.1 | ||
| HMG-CoA synthase | XM_014416338,X70034,AB733009 | HBug_USDA-ARS_IIBBL.375421 | XP_014277503.1 |
| HMG-CoA reductase | X70034,XM_014424783,XM_014391838,XM_015521221 | HBug_USDA-ARS_IIBBL.421664 | XP_014280269.1 |
| Mevalonate kinase | XM_014416757,XM_014391202,GEDC01029638,XM_012431690 | HBug_USDA-ARS_IIBBL.227208 | XP_014272243.1 |
| Phosphomevalonate kinase | XM_014416475,XM_014398812,GECZ01001991,GEBQ01010256 | HBug_USDA-ARS_IIBBL.270718 | XP_014271961.1 |
| Diphosphomevalonate decarboxylase | XM_018479978,XM_014434730,XM_014399537,GEBQ01002905 | HBug_USDA-ARS_IIBBL.428020 | XP_014290216.1 |
| IDP Isomerase | XM_014415973,XP_014247428,GECU01023093,AK417896 | HBug_USDA-ARS_IIBBL.92242 | XP_014271459.1 |
| FDP Synthase | XP_014289225 | HBug_USDA-ARS_IIBBL.420512 | XP_014276183.1 |
| HBug_USDA-ARS_IIBBL.414919 | XP_014276401.1 | ||
| Farnesyl diphosphatase | NP_572760.1 | No homologs detected | |
| Farnesol dehydrogenase | XP_014292348 | HBug_USDA-ARS_IIBBL.414590 | XP_014286519.1 |
| HBug_USDA-ARS_IIBBL.376500 | XP_014286525.1 | ||
| HBug_USDA-ARS_IIBBL.328207 | XP_014286524.1 | ||
| Farnesal dehydrogenase | KC243495 | HBug_USDA-ARS_IIBBL.79640 | XP_014292700.1 |
| HBug_USDA-ARS_IIBBL.14716 | XP_014272618.1 | ||
| Juvenile hormone acid methyltransferase | XP_014293044,XP_001651876 | HBug_USDA-ARS_IIBBL.485486 | XP_014290953.1 |
| HBug_USDA-ARS_IIBBL.519494 | XP_014293044.1 | ||
| HBug_USDA-ARS_IIBBL.346622 | XP_014283772.1 | ||
| Methyl farnesoate epoxidase | XP_014283057 | HBug_USDA-ARS_IIBBL.517163 | XP_014283057.1 |
| Harlequin Bug | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 2nd | 4th | 4th:2nd | ♂ | ♀ | ♀ : ♂ | Nymphs | Adults | Adults:Nymphs | Overall | Transcript ID |
| Acetoacetyl-CoA thiolase | 176.92 | 131.53 | -0.43 | 211.58 | 344.29 | 0.70 | 154.39 | 267.85 | 0.79 | 211.99 | HBug_USDA-ARS_IIBBL.267134 |
| Acetoacetyl-CoA thiolase | 41.01 | 28.64 | -0.52 | 21.84 | 33.13 | 0.60 | 34.61 | 26.66 | -0.38 | 30.56 | HBug_USDA-ARS_IIBBL.43170 |
| HMG-CoA reductase | 15.83 | 12.42 | -0.35 | 29.97 | 22.95 | -0.39 | 14.10 | 27.05 | 0.94 | 20.60 | HBug_USDA-ARS_IIBBL.421664 |
| HMG-CoA synthase | 0.53 | 0.60 | 0.18 | 0.27 | 0.45 | 0.74 | 0.57 | 0.35 | -0.70 | 0.45 | HBug_USDA-ARS_IIBBL.375421 |
| Mevalonate kinase | 0.11 | 0.28 | 1.35 | 0.05 | 0.33 | 2.72 | 0.22 | 0.13 | -0.76 | 0.19 | HBug_USDA-ARS_IIBBL.227208 |
| Phosphomevalonate kinase | 4.51 | 2.76 | -0.71 | 6.08 | 3.08 | -0.98 | 3.59 | 4.76 | 0.41 | 4.15 | HBug_USDA-ARS_IIBBL.270718 |
| Diphosphomevalonate decarboxylase | 0.05 | 0.12 | 1.26 | 0.13 | 0.11 | -0.24 | 0.09 | 0.14 | 0.64 | 0.15 | HBug_USDA-ARS_IIBBL.428020 |
| IDP Isomerase | 20.22 | 10.48 | -0.95 | 15.04 | 13.73 | -0.13 | 15.32 | 14.53 | -0.08 | 14.92 | HBug_USDA-ARS_IIBBL.92242 |
| FDP Synthase | 3.44 | 0.41 | -3.07 | 46.44 | 3.51 | -3.73 | 1.92 | 28.48 | 3.89 | 15.29 | HBug_USDA-ARS_IIBBL.420512 |
| FDP Synthase | 25.04 | 23.37 | -0.10 | 32.47 | 31.56 | -0.04 | 23.79 | 32.17 | 0.44 | 27.98 | HBug_USDA-ARS_IIBBL.414919 |
| Farnesol dehydrogenase | 0.16 | 0.15 | -0.09 | 0.00 | 0.00 | undefined | 0.16 | 0.00 | - ∞ | 0.08 | HBug_USDA-ARS_IIBBL.414590 |
| Farnesol dehydrogenase | 0.00 | 0.13 | + ∞ | 0.07 | 0.45 | 2.68 | 0.03 | 0.15 | 2.32 | 0.09 | HBug_USDA-ARS_IIBBL.328207 |
| Farnesol dehydrogenase | 13.27 | 9.88 | -0.43 | 7.78 | 7.54 | -0.05 | 11.57 | 7.81 | -0.57 | 9.65 | HBug_USDA-ARS_IIBBL.376500 |
| Farnesal dehydrogenase | 0.29 | 0.20 | -0.54 | 8.14 | 12.84 | 0.66 | 0.24 | 10.14 | 5.40 | 5.20 | HBug_USDA-ARS_IIBBL.14716 |
| Farnesal dehydrogenase | 3.29 | 9.14 | 1.47 | 4.99 | 5.77 | 0.21 | 6.24 | 5.30 | -0.24 | 5.78 | HBug_USDA-ARS_IIBBL.79640 |
| Juvenile hormone acid methyltransferase | 0.04 | 0.00 | - ∞ | 0.12 | 0.03 | -2.00 | 0.00 | 0.09 | + ∞ | 0.00 | HBug_USDA-ARS_IIBBL.519494 |
| Juvenile hormone acid methyltransferase | 3.04 | 10.78 | 1.83 | 4.97 | 64.51 | 3.70 | 6.87 | 30.65 | 2.16 | 17.85 | HBug_USDA-ARS_IIBBL.485486 |
| Juvenile hormone acid methyltransferase | 0.03 | 1.22 | 5.35 | 0.09 | 0.28 | 1.64 | 0.68 | 0.17 | -2.00 | 0.45 | HBug_USDA-ARS_IIBBL.346622 |
| Methyl farnesoate epoxidase | 0.39 | 0.12 | -1.70 | 0.18 | 0.14 | -0.36 | 0.25 | 0.17 | -0.56 | 0.21 | HBug_USDA-ARS_IIBBL.517163 |
| Brown Marmorated Stink Bug | |||||||||||
| Annotation | 2nd | 4th | 4th:2nd | ♂ | ♀ | ♀ : ♂ | Nymphs | Adults | Adults:Nymphs | Overall | Protein / RNA IDs |
| Acetoacetyl-CoA thiolase | 175.06 | 168.71 | -0.05 | 793.94 | 942.05 | 0.25 | 171.71 | 864.84 | 2.33 | 452.10 | XP_014294739.1 / XM_014439253.1 |
| Acetoacetyl-CoA thiolase | 70.48 | 65.24 | -0.11 | 147.12 | 185.97 | 0.34 | 67.72 | 165.71 | 1.29 | 107.36 | XP_014275331.1 / XM_014419845.1 |
| HMG-CoA reductase | 18.12 | 21.68 | 0.26 | 44.27 | 25.96 | -0.77 | 20.00 | 35.52 | 0.83 | 26.28 | XP_014280269.1 / XM_014424783.1 |
| HMG-CoA synthase | 0.00 | 0.00 | undefined | 0.44 | 1.40 | 1.67 | 0.00 | 1.28 | + ∞ | 0.54 | XP_014277503.1 / XM_014422017.1 |
| Mevalonate kinase | 8.00 | 8.54 | 0.09 | 12.13 | 7.82 | -0.63 | 8.29 | 10.07 | 0.28 | 9.01 | XP_014272243.1 / XM_014416757.1 |
| Phosphomevalonate kinase | 5.97 | 4.46 | -0.42 | 7.02 | 4.80 | -0.55 | 5.17 | 5.96 | 0.21 | 5.49 | XP_014271961.1 / XM_014416475.1 |
| Diphosphomevalonate decarboxylase | 3.96 | 3.44 | -0.20 | 4.90 | 3.98 | -0.30 | 3.72 | 4.46 | 0.26 | 4.04 | XP_014290216.1 / XM_014434740.1 |
| IDP Isomerase | 13.51 | 10.56 | -0.36 | 11.89 | 14.15 | 0.25 | 11.96 | 12.97 | 0.12 | 12.37 | XP_014271459.1 / XM_014415973.1 |
| FDP Synthase | 3.62 | 8.08 | 1.16 | 15.70 | 18.66 | 0.25 | 5.97 | 17.12 | 1.52 | 10.48 | XP_014276183.1 / XM_014420697.1 |
| FDP Synthase | 12.52 | 14.96 | 0.26 | 13.13 | 15.78 | 0.27 | 13.80 | 14.40 | 0.06 | 14.04 | XP_014276401.1 / XM_014420915.1 |
| FDP synthase | 0.04 | 0.16 | 2.00 | 0.80 | 0.04 | -4.32 | 0.10 | 0.44 | 2.14 | 0.24 | XP_014289203.1 / XM_014433717.1 |
| FDP synthase | 0.29 | 0.29 | 0.00 | 0.35 | 0.22 | -0.67 | 0.29 | 0.28 | -0.05 | 0.29 | XP_014289225.1 / XM_014433739.1 |
| Farnesol dehydrogenase | 204.63 | 77.01 | -1.41 | 35.70 | 46.26 | 0.37 | 137.30 | 40.68 | -1.75 | 98.17 | XP_014286519.1 / XM_014431033.1 |
| Farnesol dehydrogenase | 78.56 | 47.27 | -0.73 | 43.79 | 43.94 | 0.00 | 62.05 | 43.86 | -0.50 | 54.69 | XP_014286524.1 / XM_014431038.1 |
| Farnesol dehydrogenase | 107.93 | 53.10 | -1.02 | 44.15 | 27.32 | -0.69 | 79.01 | 36.10 | -1.13 | 61.65 | XP_014286525.1 / XM_014431039.1 |
| Farnesal dehydrogenase | 28.66 | 94.12 | 1.72 | 35.72 | 49.72 | 0.48 | 63.19 | 42.42 | -0.57 | 54.79 | XP_014272618.1 / XM_014417132.1 |
| Farnesal dehydrogenase | 49.42 | 54.55 | 0.14 | 57.72 | 65.03 | 0.17 | 52.13 | 61.22 | 0.23 | 55.81 | XP_014292700.1 / XM_014437214.1 |
| Juvenile hormone acid methyltransferase | 0.13 | 0.06 | -1.12 | 0.22 | 34.99 | 7.31 | 0.09 | 16.85 | 7.55 | 6.87 | XP_014293044.1 / XM_014437558.1 |
| Juvenile hormone acid methyltransferase | 46.90 | 45.31 | -0.05 | 22.24 | 79.28 | 1.83 | 46.06 | 49.52 | 0.10 | 47.46 | XP_014290953.1 / XM_014435467.1 |
| Juvenile hormone acid methyltransferase | 0.38 | 0.34 | -0.16 | 0.79 | 0.18 | -2.13 | 0.36 | 0.50 | 0.47 | 0.41 | XP_014283772.1 / XM_014428286.1 |
| Methyl farnesoate epoxidase | 1.15 | 0.59 | -0.96 | 0.16 | 0.26 | 0.70 | 0.86 | 0.21 | -2.03 | 0.59 | XP_014283057.1 / XM_014427571.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparks, M.E.; Rhoades, J.H.; Nelson, D.R.; Kuhar, D.; Lancaster, J.; Lehner, B.; Tholl, D.; Weber, D.C.; Gundersen-Rindal, D.E. A Transcriptome Survey Spanning Life Stages and Sexes of the Harlequin Bug, Murgantia histrionica. Insects 2017, 8, 55. https://doi.org/10.3390/insects8020055
Sparks ME, Rhoades JH, Nelson DR, Kuhar D, Lancaster J, Lehner B, Tholl D, Weber DC, Gundersen-Rindal DE. A Transcriptome Survey Spanning Life Stages and Sexes of the Harlequin Bug, Murgantia histrionica. Insects. 2017; 8(2):55. https://doi.org/10.3390/insects8020055
Chicago/Turabian StyleSparks, Michael E., Joshua H. Rhoades, David R. Nelson, Daniel Kuhar, Jason Lancaster, Bryan Lehner, Dorothea Tholl, Donald C. Weber, and Dawn E. Gundersen-Rindal. 2017. "A Transcriptome Survey Spanning Life Stages and Sexes of the Harlequin Bug, Murgantia histrionica" Insects 8, no. 2: 55. https://doi.org/10.3390/insects8020055
APA StyleSparks, M. E., Rhoades, J. H., Nelson, D. R., Kuhar, D., Lancaster, J., Lehner, B., Tholl, D., Weber, D. C., & Gundersen-Rindal, D. E. (2017). A Transcriptome Survey Spanning Life Stages and Sexes of the Harlequin Bug, Murgantia histrionica. Insects, 8(2), 55. https://doi.org/10.3390/insects8020055