Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures

{kind=link}
{kind=link}
Abstract
:1. Introduction
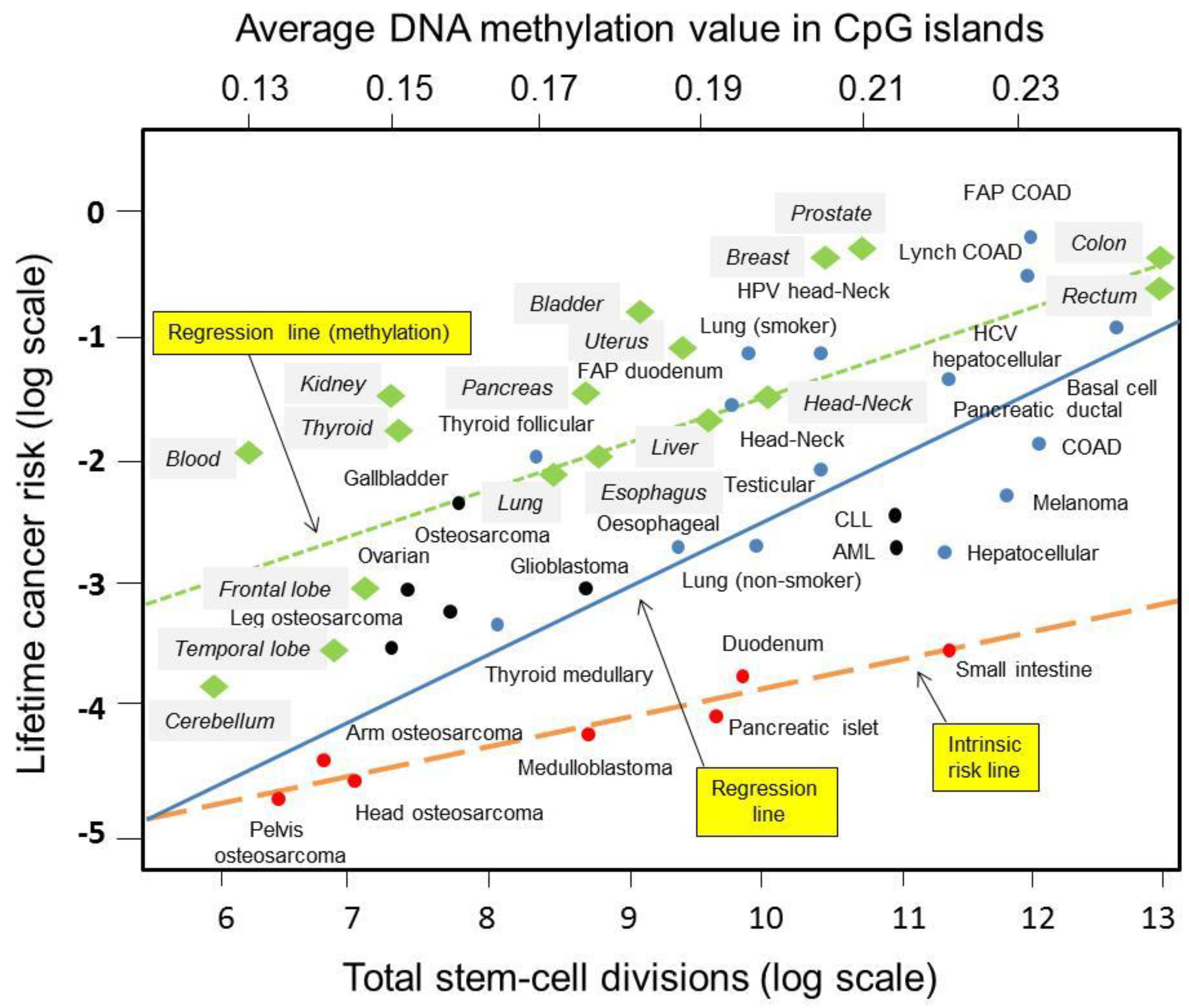
2. Tomasetti and Vogelstein Hypothesis
3. Stem Cells and Cancer
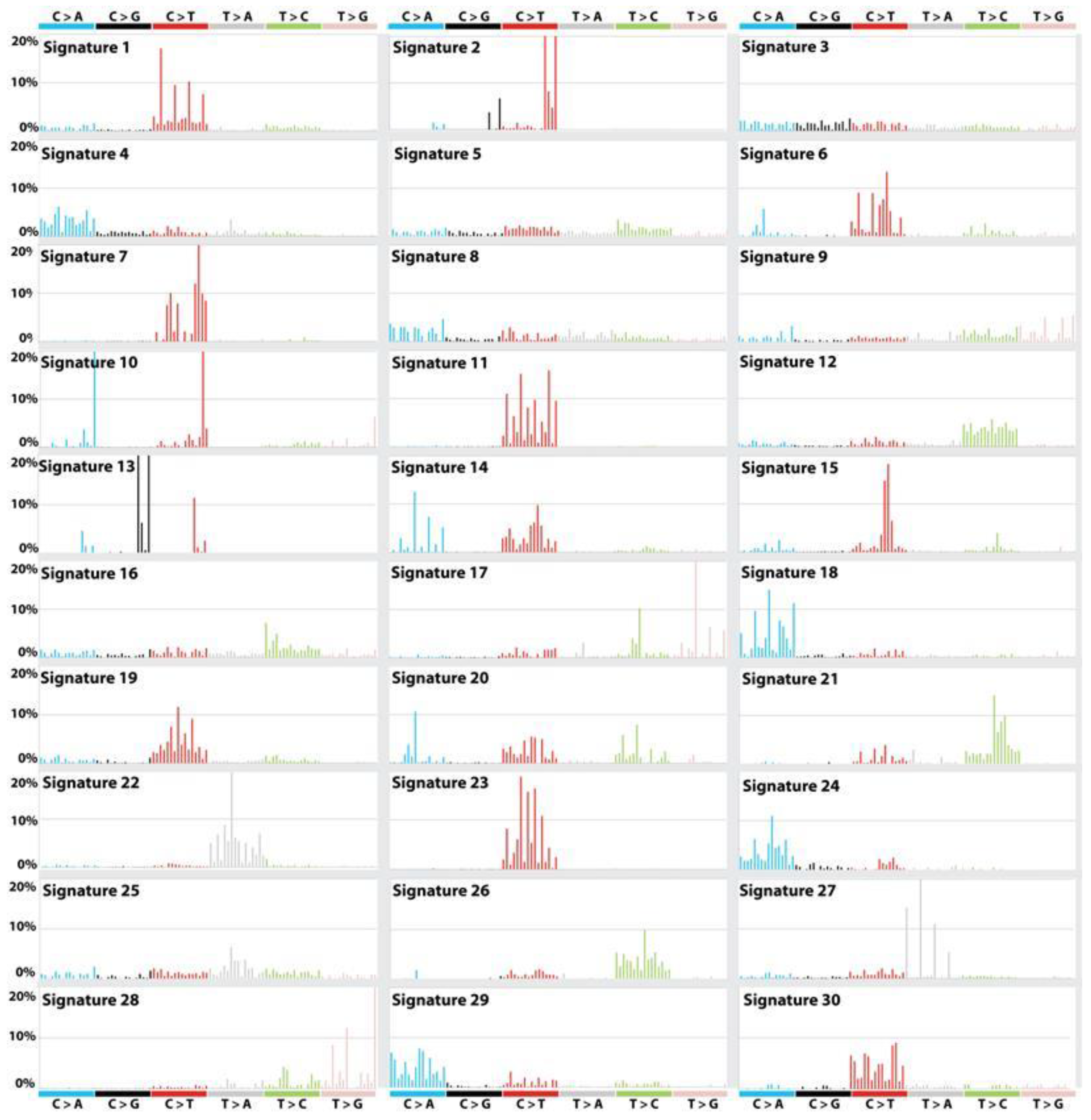
4. DNA Repair and Cancer Stem Cells
5. Cancer Stem Cell and Epigenomic Reprogramming
6. Discussion and Perspectives
Funding
Conflicts of Interest
References
- Egeblad, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [Green Version]
- Navin, N.E.; Hicks, J. Tracing the tumor lineage. Mol. Oncol. 2010, 4, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.F.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peto, R.; Roe, F.J.C.; Lee, P.N.; Levy, L.; Clack, J. Cancer and ageing in mice and men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunney, L.; Muir, B. Peto’s paradox and the hallmarks of cancer: Constructing an evolutionary framework for understanding the incidence of cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150161. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, A.S. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum. Mutat. 2013, 21, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pazhanisamy, S.K. Stem cells, DNA damage, ageing and cancer. Hematol. Oncol. Stem Cell Ther. 2009, 2, 375–384. [Google Scholar] [CrossRef]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2008, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, P.; Shibata, D. A simple algebraic cancer equation: Calculating how cancers may arise with normal mutation rates. BMC Cancer 2010, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrret, R.; Moseley, S. Evolution of resistance and progression to disease during clonal expansion of cancer. Theor. Popul. Biol. 2010, 77, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddel, R.R. The role of senescence and immortalization in carcinogenesis. Carcinogenesis 2000, 21, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Furth, J.; Kahn, M. The transmission of leukaemia of mice with a single cell. Am. J. Cancer 1937, 31, 276–282. [Google Scholar]
- Wang, J.C.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Block, C.; Georg, T.I.; Galli, S.J.; Gotlib, J.; Haferlach, T.; Hoermann, G.; Hermine, O.; et al. Proposed terminology and classification of pre-malignant neoplastic conditions: A consensus proposal. EBioMedicine 2017, 16, 17–24. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Smith, B.D. Eliminating cancer stem cells by targeting embryonic signaling pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Weeden, C.E.; Asselin-Labat, M.L. Mechanisms of DNA damage repair in adult stem cells and implications for cancer formation. Biochim. Biophys. Acta 2018, 1864, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.; Lerner, L.K.; Okamoto, O.K.; Marchetto, M.C.; Menck, C.F. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. 2013, 752, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Altrock, P.M.; Liu, L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 12, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Rabhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.P.; Sproul, D.; Meehan, R.R. DNA methylation reprogramming in cancer: Does it act by re-configuring the binding landscape of Polycomb repressive complexes? Bioessays 2014, 36, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.P.; Rao, A. DNA methylation and methylcytosine oxidation in cell fate decisions. Curr. Opin. Cell Biol. 2013, 25, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klutstein, M.; Moss, J.; Kaplan, M.; Cedar, H. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc. Natl. Acad. Sci. USA 2017, 114, 2230–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1981, 166, 1191–1308. [Google Scholar] [CrossRef]
- Blot, W.L.; Tarone, R.E. Doll and Peto’s quantitative estimates of cancer risks: Holding generally true for 35 years. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.V. Cancer: Bad luck or punishment? Biochemistry (Moscow) 2017, 82, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Waclaw, B. Genes, environment, and ‘‘bad luck’’. Science 2017, 355, 1266–1267. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.; Kaltz, O.; Hochberg, M.E. Peto’s paradox and human cancers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150104. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G.; Belshaw, R.; Katzourakis, A. “There and back again”: Revisiting the pathophysiological roles of human endogenous retroviruses in the post-genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120504. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Jacobs, D.R., Jr.; Porta, M. Hypothesis: A unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ. Health Perspect. 2009, 117, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.E.; Sangiuliano, B.A.; Perez-Sosa, M.; Neyra, J.M.; Moreira, D.F. Using pharmacogenomic databases for discovering patient-target genes and small molecule candidates to cancer therapy. Front. Pharmacol. 2016, 7, 312. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belizário, J.E. Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures. Epigenomes 2018, 2, 13. https://doi.org/10.3390/epigenomes2030013
Belizário JE. Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures. Epigenomes. 2018; 2(3):13. https://doi.org/10.3390/epigenomes2030013
Chicago/Turabian StyleBelizário, José E. 2018. "Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures" Epigenomes 2, no. 3: 13. https://doi.org/10.3390/epigenomes2030013
APA StyleBelizário, J. E. (2018). Cancer Risks Linked to the Bad Luck Hypothesis and Epigenomic Mutational Signatures. Epigenomes, 2(3), 13. https://doi.org/10.3390/epigenomes2030013