On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells



Abstract
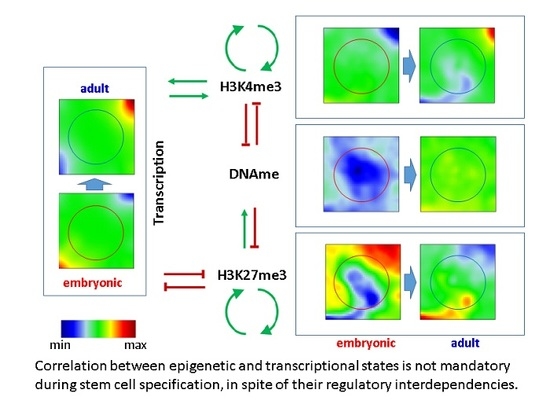
:
1. Introduction
2. Results
2.1. Transcription ‘Dominated’ Self-Organizing Maps (SOMs)
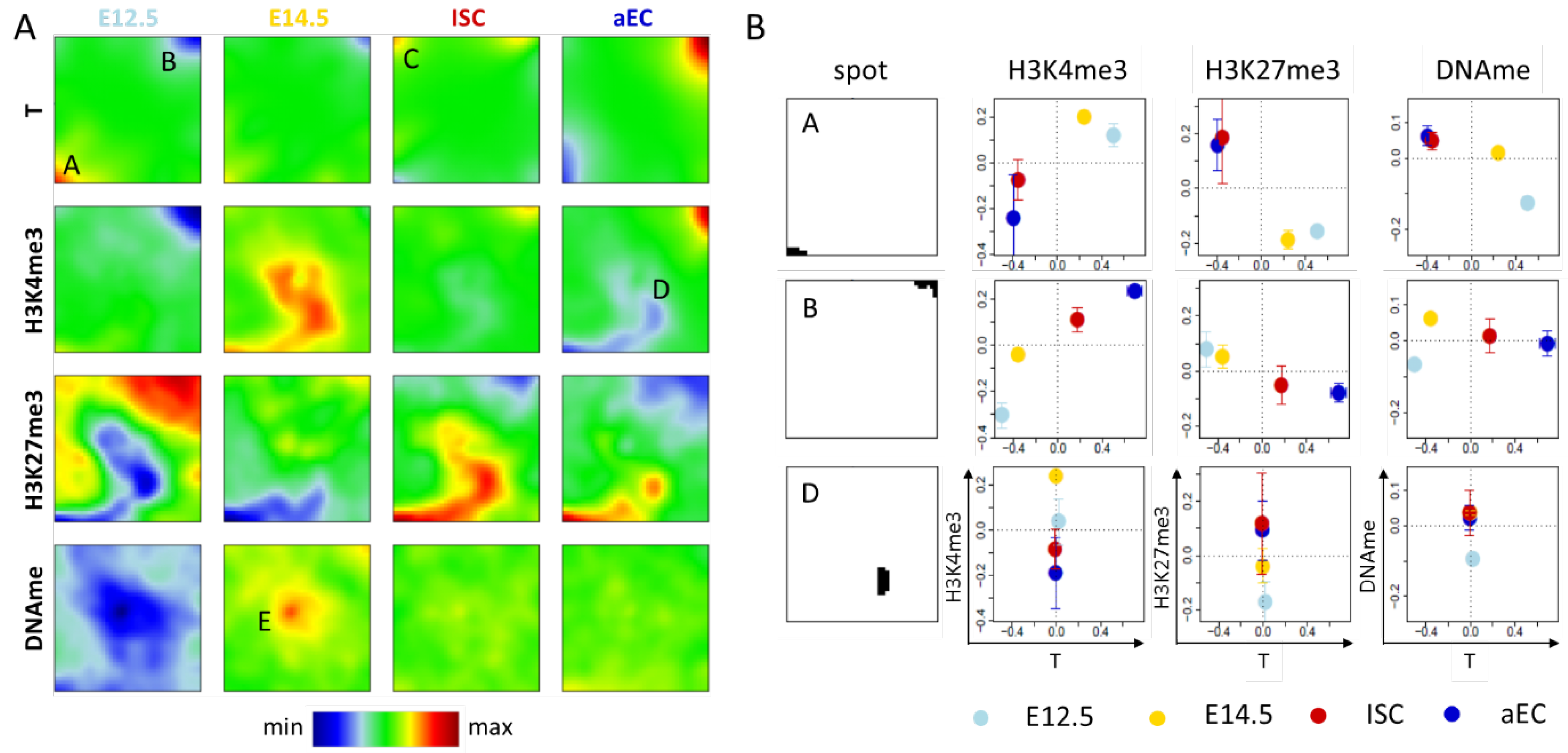
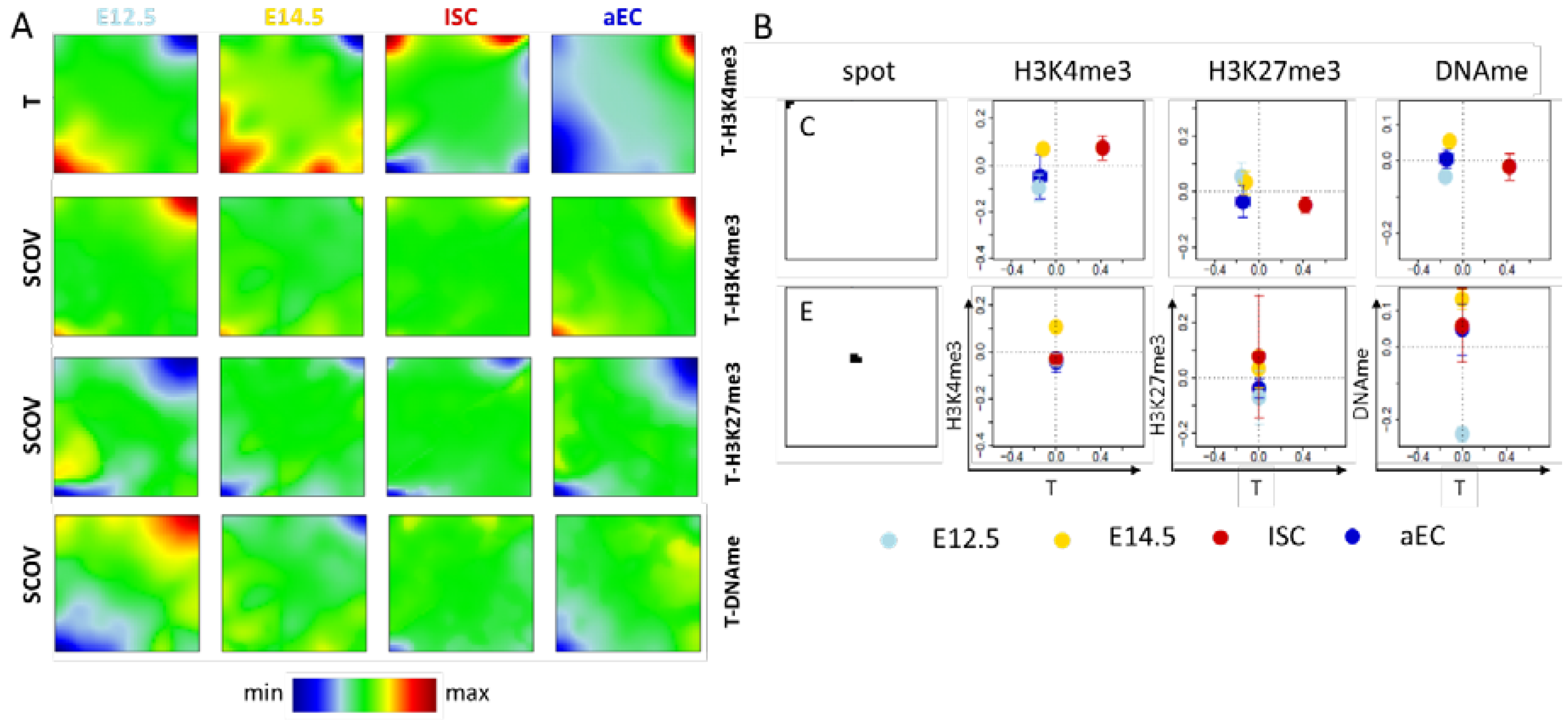
2.1.1. Correlated Changes of Epigenetic Profiles and Transcription
2.1.2. Epigenetic Regulation without Changes of Transcription
2.2. SOMs ‘Dominated’ by Epigenetics
2.2.1. H3K4me3 ‘Dominated’ SOMs
2.3. Model Considerations
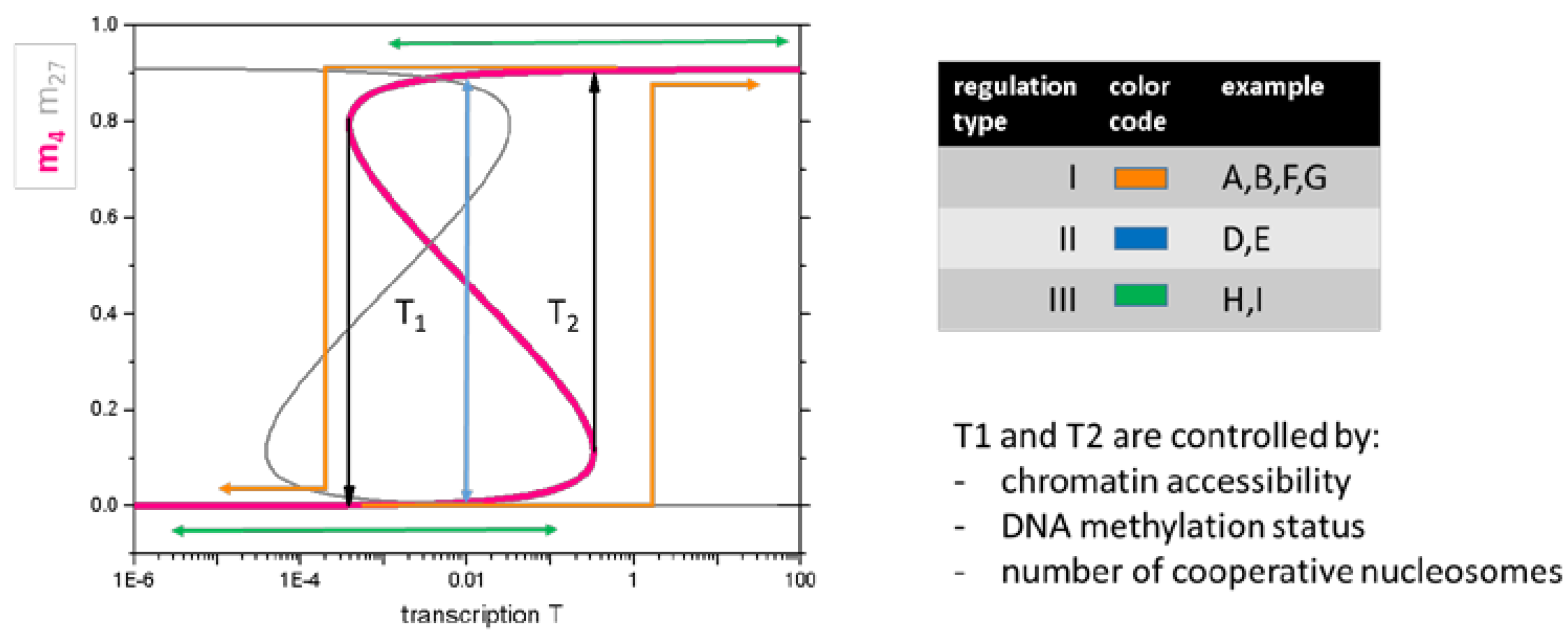
2.3.1 Regulation of Histone Modifications
2.3.2. Regulation of Transcription
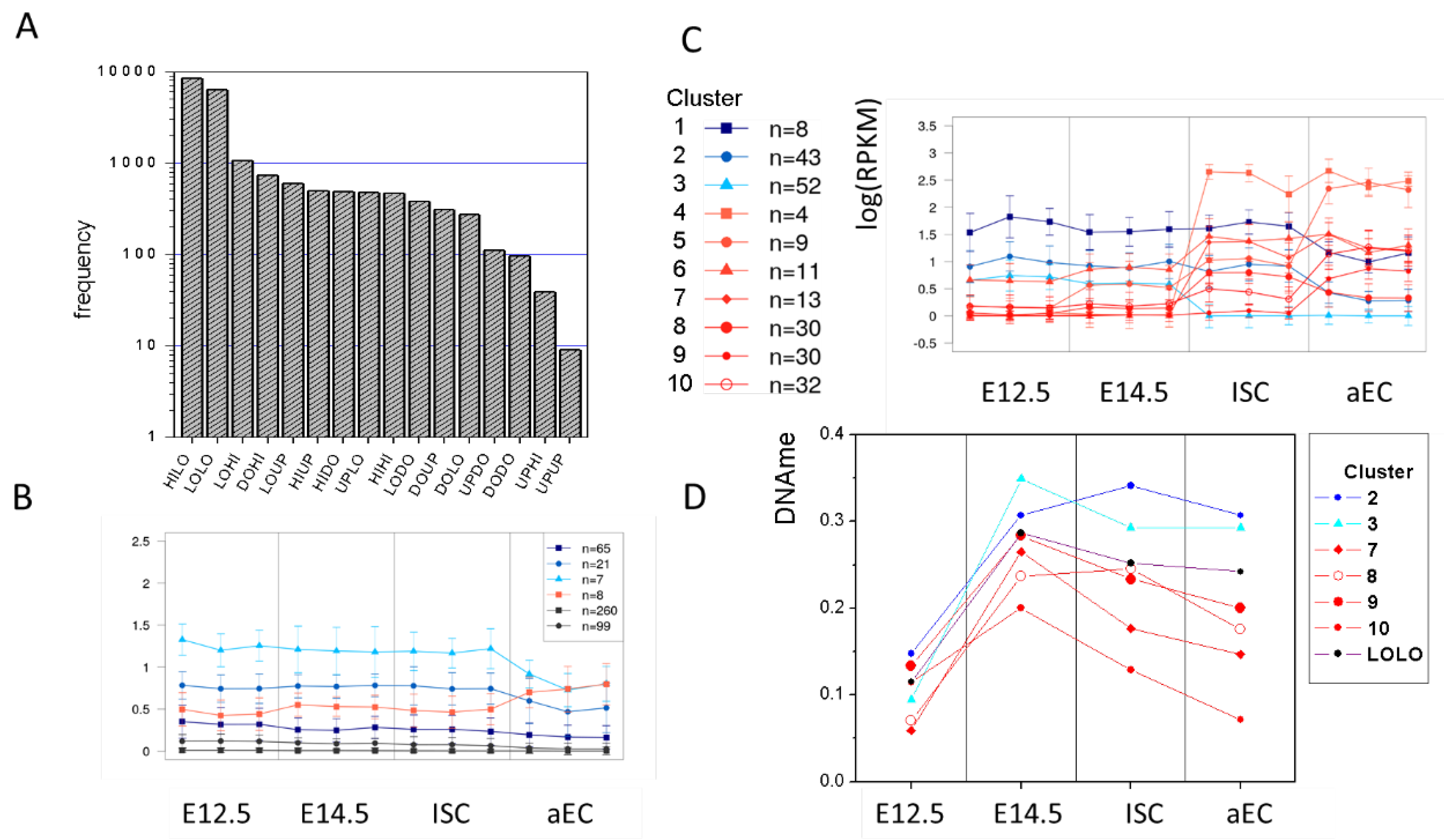
2.4. Epigenetic Regulation of Transcription: Transition States
2.4.1. Properties of Transition States
2.4.2. Clustering of State Specific Transcription Profiles
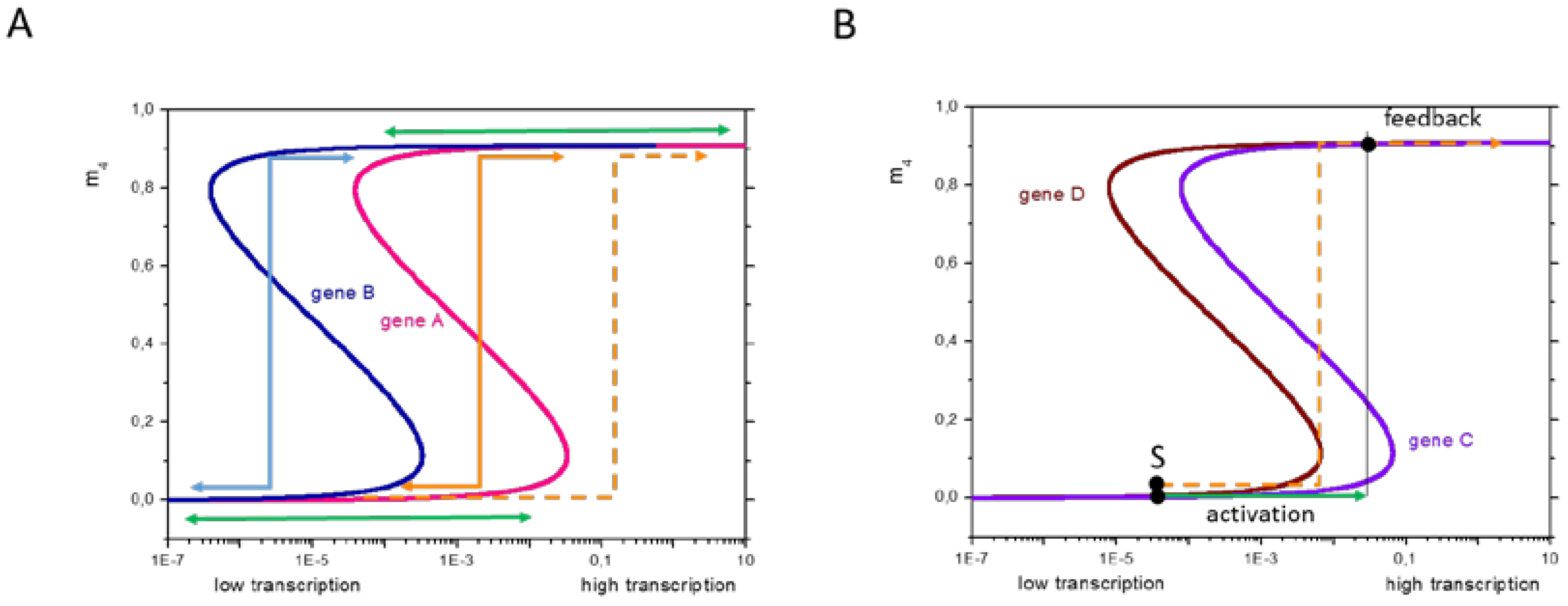
2.5. TF-Based Regulation of Transcription: Adaptive States
2.5.1. Properties of Adaptive States
2.5.2. Regulation Types Are Associated with Specific Promotor Types
3. Discussion
4. Materials and Methods
4.1. Data and Preprocessing for SOM
4.2. Combinatorial SOM Method and Gene Annotation
4.3. Identification of Transition States and Clustering of Transcriptional Profiles
Author Contributions
Funding
Conflicts of Interest
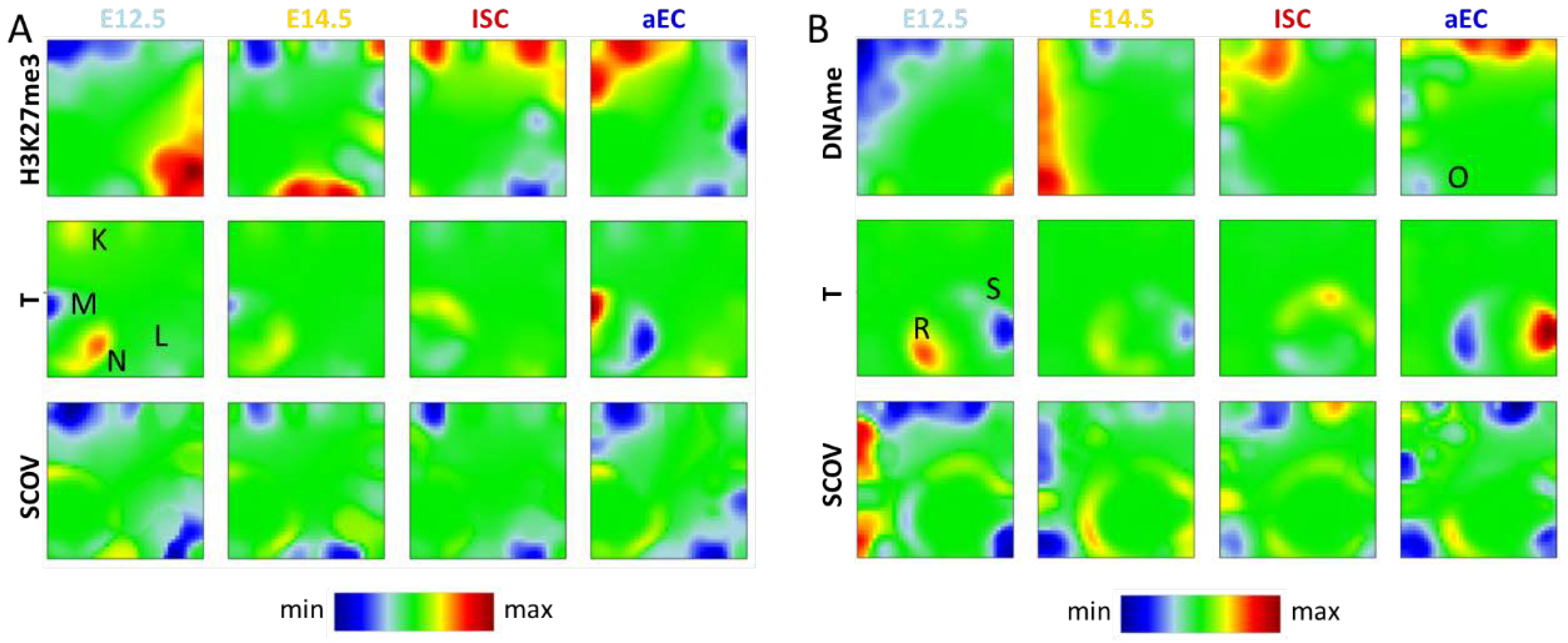
Appendix A. Additional SOM Analysis



Appendix B. Algorithm to Cluster Transcriptional Profiles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Value |
|---|---|---|
| dmin | Minimum distance (least square method) to assign a gene to a cluster | 1.8 (default) 3 (HI-LO, LO-LO) |
| dinit | Minimum distance to all available clusters to create a new cluster | 1.5 × dmin |
| imax | Max number of iterations | 250 |
| nC | Minimum number of genes in a cluster | 3 |

References
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, K.; Zang, C.; Roh, T.Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Malatesta, M.; Jung, H.R.; Walfridsson, J.; Willer, A.; Olsson, L.; Skotte, J.; Wutz, A.; Porse, B.; Jensen, O.N.; et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010, 38, 4958–4969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratowski, S.; Kim, T. The role of cotranscriptional histone methylations. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Jermann, P.; Hoerner, L.; Burger, L.; Schübeler, D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3415–E3421. [Google Scholar] [CrossRef] [PubMed]
- Mutskov, V.; Felsenfeld, G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 2004, 23, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Bintu, L.; Yong, J.; Antebi, Y.E.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, M.; Appanah, R.; Lee, S.; Lam, L.L.; Goyal, P.; Lorincz, M.C. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009, 4, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Steiner, L.; Przybilla, J.; Rohlf, T.; Prohaska, S.; Galle, J. Transcriptional regulation by histone modifications, towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 2013, 10, 026006. [Google Scholar] [CrossRef] [PubMed]
- Thalheim, T.; Herberg, M.; Loeffler, M.; Galle, J. The Regulatory Capacity of Bivalent Genes—A Theoretical Approach. Int. J. Mol. Sci. 2017, 18, 1069. [Google Scholar] [CrossRef] [PubMed]
- Hamidouche, Z.; Rother, K.; Przybilla, J.; Krinner, A.; Clay, D.; Hopp, L.; Fabian, C.; Stolzing, A.; Binder, H.; Charbord, P.; et al. Bistable Epigenetic States Explain Age-Dependent Decline in Mesenchymal Stem Cell Heterogeneity. Stem Cells 2017, 35, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Rohlf, T.; Loeffler, M.; Galle, J. Understanding epigenetic changes in aging stem cells—A computational model approach. Aging Cell 2014, 13, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Hopp, L.; Lübbert, M.; Loeffler, M.; Galle, J. Targeting DNA hypermethylation, Computational modeling of DNA demethylation treatment of acute myeloid leukemia. Epigenetics 2017, 12, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Thalheim, T.; Herberg, M.; Galle, J. Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine. Genes (Basel) 2018, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Steiner, L.; Hopp, L.; Wirth, H.; Galle, J.; Binder, H.; Prohaska, S.J.; Rohlf, T. A global genome segmentation method for exploration of epigenetic patterns. PLoS ONE 2012, 7, e46811. [Google Scholar] [CrossRef] [PubMed]
- Hopp, L.; Löffler-Wirth, H.; Galle, J.; Binder, H. Combined SOM-portrayal of transcription and DNA methylation landscapes disentangles modes of epigenetic regulation in glioblastoma. Epigenomics 2018, 10, 745–764. [Google Scholar] [CrossRef] [PubMed]
- Kazakevych, J.; Sayols, S.; Messner, B.; Krienke, C.; Soshnikova, N. Dynamic changes in chromatin states during specification and differentiation of adult intestinal stem cells. Nucleic Acids Res. 2017, 45, 5770–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H., 3rd; Schones, D.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11, expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Wirth, H.; Galle, J. Gene expression density profiles characterize modes of genomic regulation: Theory and experiment. J. Biotechnol. 2010, 149, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Wang, H.; John, S.; Shafer, A.; Canfield, T.; Lee, K.; Stamatoyannopoulos, J.A. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015, 12, 1184–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elango, N.; Yi, S.V. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol. Biol. Evol. 2008, 25, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Gates, LA.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’, Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Tsankov, A.M.; Gu, H.; Akopian, V.; Ziller, M.J.; Donaghey, J.; Amit, I.; Gnirke, A.; Meissner, A. Transcription factor binding dynamics during human ES cell differentiation. Nature 2015, 518, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Tan, Y.; Huang, T.; Ding, G.; Tu, Z.; Liu, L.; Li, Y.; Dai, H.; Xie, L. TF-centered downstream gene set enrichment analysis, Inference of causal regulators by integrating TF-DNA interactions and protein post-translational modifications information. BMC Bioinform. 2010, 11 (Suppl. 11), S5. [Google Scholar] [CrossRef] [PubMed]
- Budden, D.M.; Hurley, D.G.; Cursons, J.; Markham, J.F.; Davis, M.J.; Crampin, E.J. Predicting expression, the complementary power of histone modification and transcription factor binding data. Epigenetics Chromatin 2014, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bucher, P. Predicting transcription factor site occupancy using DNA sequence intrinsic and cell-type specific chromatin features. BMC Bioinform. 2016, 17 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Relton, C.L. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018, 19, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Ecker, S.; Pancaldi, V.; Valencia, A.; Beck, S.; Paul, D.S. Epigenetic and Transcriptional Variability Shape Phenotypic Plasticity. Bioessays 2018, 40, 1700148. [Google Scholar] [CrossRef] [PubMed]
- Loeffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on Bioconductor. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, J.B. Some Methods for classification and Analysis of Multivariate Observations. In Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; pp. 281–297. [Google Scholar]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thalheim, T.; Hopp, L.; Binder, H.; Aust, G.; Galle, J. On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes 2018, 2, 20. https://doi.org/10.3390/epigenomes2040020
Thalheim T, Hopp L, Binder H, Aust G, Galle J. On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes. 2018; 2(4):20. https://doi.org/10.3390/epigenomes2040020
Chicago/Turabian StyleThalheim, Torsten, Lydia Hopp, Hans Binder, Gabriela Aust, and Joerg Galle. 2018. "On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells" Epigenomes 2, no. 4: 20. https://doi.org/10.3390/epigenomes2040020
APA StyleThalheim, T., Hopp, L., Binder, H., Aust, G., & Galle, J. (2018). On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes, 2(4), 20. https://doi.org/10.3390/epigenomes2040020