Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance

 and
and 
Abstract
:1. Introduction
2. Results
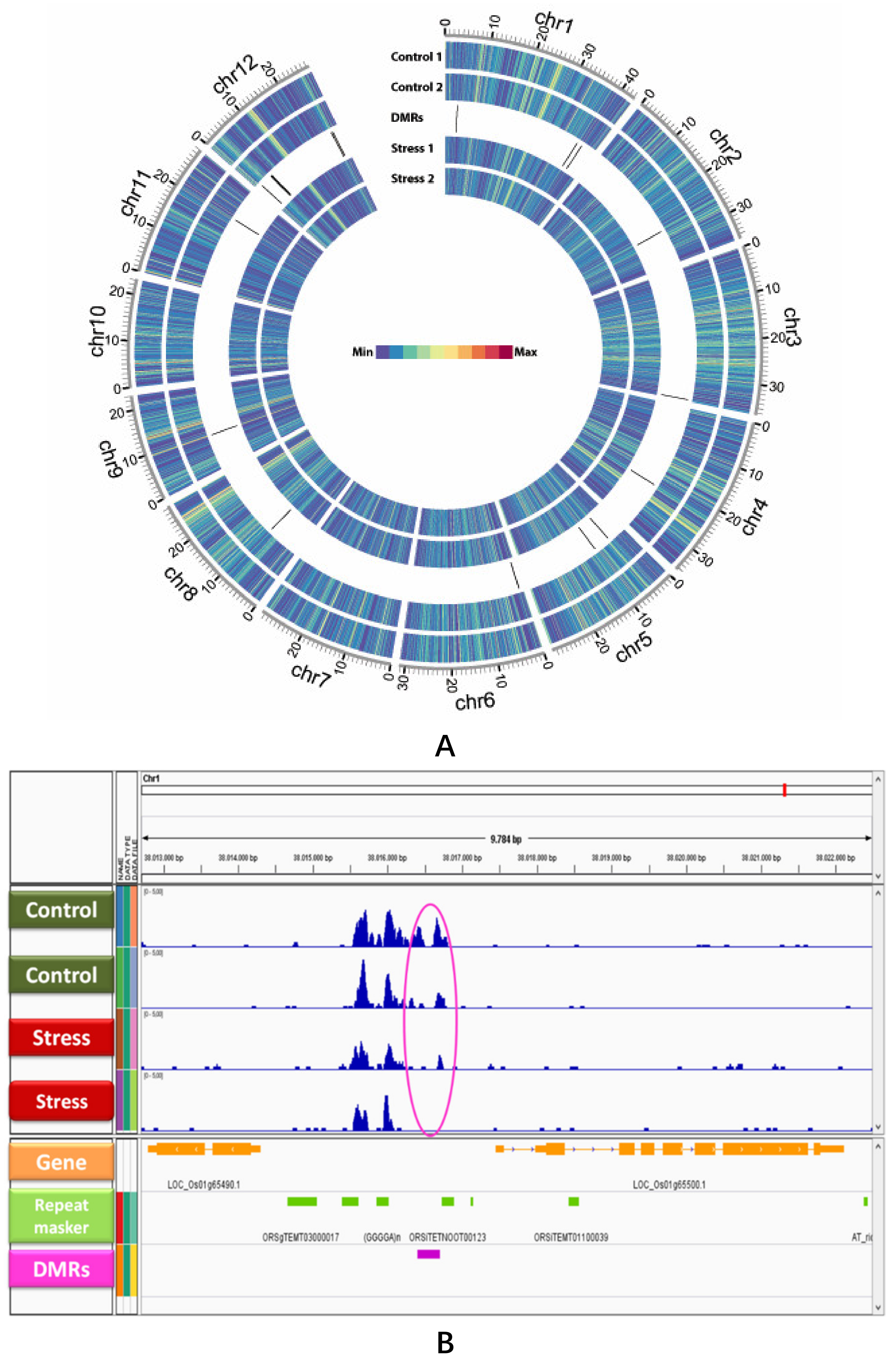
2.1. The ‘Pokkali’ Methylome and the Identification of DMRs between Salinity and Control
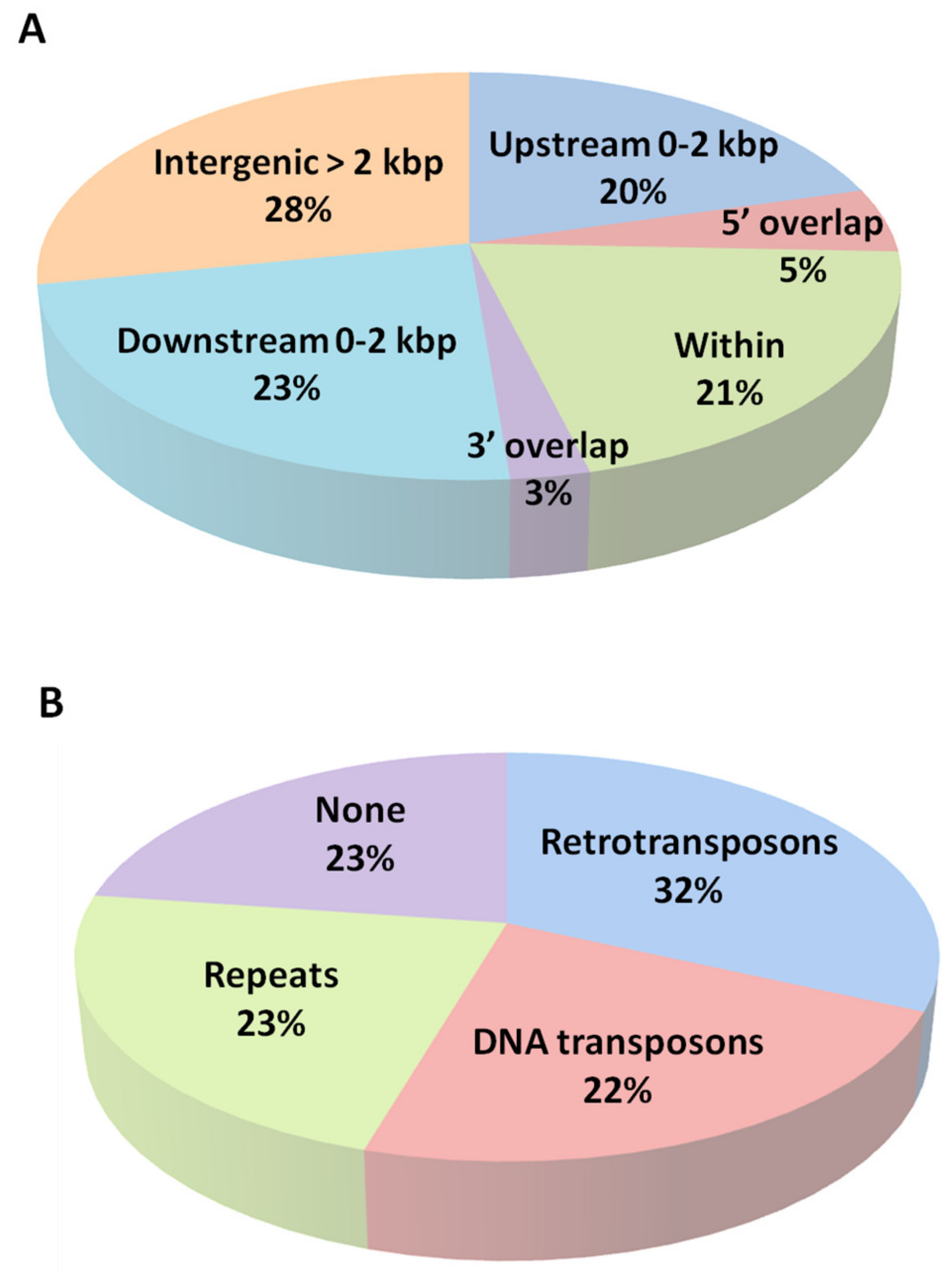
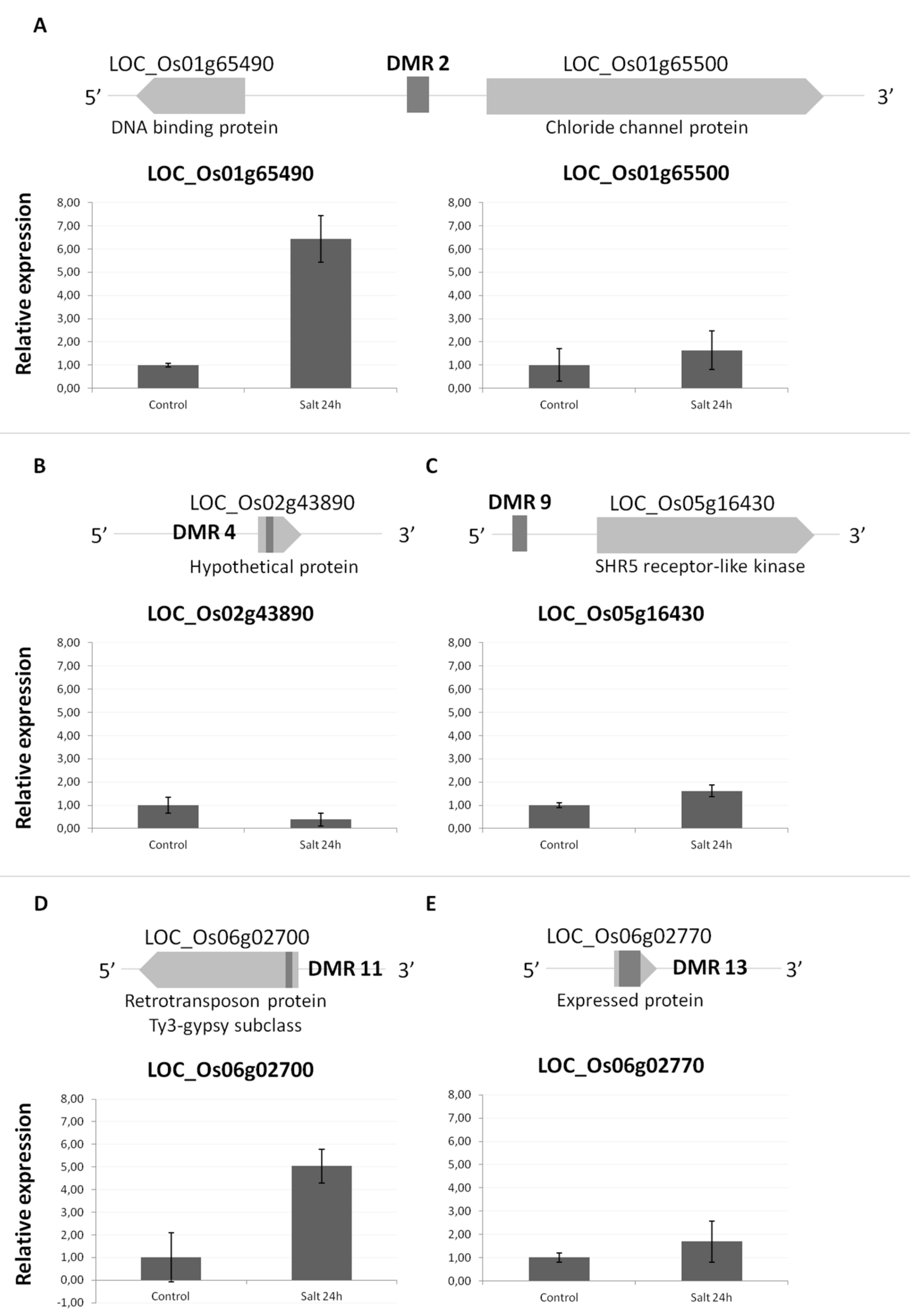
2.2. Location of DMRs Might Influence Regulation of Genes Nearby
3. Discussion
4. Materials and Methods
4.1. Plant Material, Growth Conditions, and Salt Stress Treatment
4.2. Methylated DNA Immunoprecipitation Sequencing (MeDIP-Seq)
4.3. Mapping and Processing the MeDIP-Seq Reads
4.4. Identification of Differentially Methylated Regions (DMRs)
4.5. Bisulfite Sequencing (BS) of DMRs
4.6. Gene Expression Studies by Real-Time RT-PCR
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Jagendorf, A.; Zhu, J.K. Understanding and improving salt tolerance in plants. Crop Sci. 2005, 45, 437–448. [Google Scholar] [CrossRef]
- Ghosh, B.; Ali Md, N.; Saikat, G. Response of Rice under Salinity Stress: A Review Update. J. Res. Rice 2016, 4, 167. [Google Scholar] [CrossRef]
- Kumar, K.; Kumar, M.; Kim, S.R.; Ryu, H.; Cho, Y.G. Insights into genomics of salt stress response in rice. Rice 2013, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Choi, J.; An, G.; Kim, S.R. Ectopic Expression of OsSta2 enhances salt stress tolerance in rice. Front. Plant Sci. 2012, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sang-Choon, L.; Ji-Youn, K.; Soo-Jin, K.; San, S.A.; Seong-Ryong, K. Over-expression of Dehydrin Gene, OsDhn1, improves drought and salt stress tolerance through scavenging of reactive oxygen species in rice (Oryza sativa L.). J. Plant Biol. 2014, 57, 383–393. [Google Scholar] [CrossRef]
- Xie, R.; Zhang, J.; Ma, Y.; Pan, X.; Dong, C.; Pang, S.; He, S.; Deng, L.; Yi, S.; Zheng, Y.; et al. Combined analysis of mRNA and miRNA identifies dehydration and salinity responsive key molecular players in citrus roots. Sci. Rep. 2017, 6, 42094. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, M.; Zhao, Y.; He, X.; Ding, C.; Wang, S.; Feng, Y.; Song, X.; Li, P.; Wang, B. Identification of salt tolerance-related microRNAs and their targets in maize (Zea mays L.) using high-throughput sequencing and degradome analysis. Front. Plant Sci. 2017, 26, 864. [Google Scholar] [CrossRef]
- Santos, A.P.; Ferreira, L.; Maroco, J.; Oliveira, M.M. Abiotic stress and induced DNA hypomethylation cause interphase chromatin structural changes in rice rDNA loci. Cytogenet. Genome Res. 2011, 132, 297–303. [Google Scholar] [CrossRef]
- Santos, A.P.; Ferreira, L.J.; Oliveira, M.M. Concerted flexibility of chromatin structure, methylome and histone modifications along with plant stress responses. Biology 2017, 6, 3. [Google Scholar] [CrossRef]
- Wang, W.S.; Zhao, X.Q.; Pan, Y.; Zhu, L.H.; Fu, B.Y.; Li, Z.K. DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. J. Genet. Genom. 2011, 38, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef] [PubMed]
- Hashida, S.N.; Kitamura, K.; Mikami, T.; Kishima, Y. Temperature shift coordinately changes the activity and the methylation state of transposon Tam3 in Antirrhinum majus. Plant Physiol. 2003, 132, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Feng, S.J.; Zhang, J.J.; Luo, F.; Zhang, S.; Yang, H. Genome-wide identification of DNA methylation provides insights into the association of gene expression in rice exposed to pesticide atrazine. Sci. Rep. 2016, 6, 18985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.P.; Abranches, R.; Alison, E.; Stoger, E.; Viegas, W.; Shaw, P.J. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J. Cell Sci. 2002, 115, 4597–4606. [Google Scholar] [CrossRef]
- Wang, M.; Qin, L.M.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Qin, Q.; Sun, F.; Wang, Y.; Xu, D.; Li, Z.; Fu, B. Genome-Wide Differences in DNA Methylation Changes in Two Contrasting Rice Genotypes in Response to Drought Conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef]
- Garg, R.; Chevala, V.V.S.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef]
- Pandey, G.; Yada, C.B.; Sahu, P.P.; Muthamilarasan, M.; Prasad, M. Salinity induced differential methylation patterns in contrasting cultivars of foxtail millet (Setaria italica L.). Plant Cell Rep. 2017, 36, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Wang, M.C.; Xia, G.M. A transcriptomic analysis reveals the nature of salinity tolerance of a wheat introgression line. Plant Mol. Biol. 2012, 78, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Comai, L.; Tyagi, A.P.; Winter, K.; Holmes-Davis, R.; Reynolds, S.H.; Stevens, Y.; Byers, B. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 2000, 12, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Smith, J.F.; Kimura, M.T.; Morrow, A.D.; Matsuyama, T.; Nagase, H.; Held, W.A. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. PNAS 2005, 102, 9, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Thorne, N.P.; Flicek, P.; Kulesha, E.; Gräf, S.; Tomazou, E.M.; Bäckdahl, L.; Johnson, N.; Herberth, M.; et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008, 18, 1518–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slieker, R.C.; Bos, S.D.; Goeman, J.J.; Bovée, J.V.; Talens, R.P.; van der Breggen, R.; Suchiman, H.E.; Lameijer, E.W.; Putter, H.; van den Akker, E.B.; et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenet. Chromatin 2013, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49. [Google Scholar] [CrossRef]
- Vining, K.; Pomraning, K.R.; Wilhelm, L.J.; Ma, C.; Pellegrini, M.; Di, Y.; Mockler, T.C.; Freitag, M.; Strauss, S.H. Methylome reorganization during in vitro dedifferentiation and regeneration of Populus trichocarpa. BMC Plant Biol. 2013, 13, 92. [Google Scholar] [CrossRef]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 2013, 8, 2783–2797. [Google Scholar] [CrossRef]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaneri, A.C.; Jones, A.M. Genome-wide quantitative identification of DNA differentially methylated sites in Arabidopsis seedlings growing at different water potential. PLoS ONE 2013, 8, e59878. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, L.; Li, M.; Lou, Q.; Xia, H.; Wang, P.; Li, T.; Liu, H.; Luo, L. Transgenerational variations in DNA methylation induced by drought stress in two rice varieties with distinguished difference to drought resistance. PLoS ONE 2013, 8, e80253. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.; Tomazou, E.M.; Brinkman, A.B.; Müller, F.; Simmer, F.; Gu, H.; Jäger, N.; Gnirke, A.; Stunnenberg, H.G.; Meissner, A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol. 2010, 28, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Yong, W.-S.; Hsu, F.-M.; Chen, P.-Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Nagaki, K.; Cheng, Z.; Ouyang, S.; Talbert, P.B.; Kim, M.; Jones, K.M.; Henikoff, S.; Buell, C.R.; Jian, J. Sequencing of a rice centromere uncovers active genes. Nat. Genet. 2004, 36, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Kikuchi, S.; Yan, H.; Zhang, W.; Rosenbaum, H.; Iniguez, A.L.; Jiang, J. Euchromatic subdomains in rice centromeres are associated with genes and transcription. Plant Cell 2011, 23, 4054–4064. [Google Scholar] [CrossRef]
- Santos, A.P.; Shaw, P.J. Interphase chromosomes and the Rabl configuration: Does genome size matter? J. Microsc. 2004, 214, 201–206. [Google Scholar] [CrossRef]
- Fransz, P.; de Jong, J.H.; Lysak, M.; Castiglione, M.R.; Schubert, I. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. PNAS 2002, 99, 14584–14589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, W.; Fischer, T.C.; Winderl, S.; Fransz, P.; Torres-Ruiz, R.A. The centromere1 (CEN1) region of Arabidopsis thaliana: Architecture and functional impact of chromatin. Plant J. 2001, 27, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.F.; Zhu, J.K. RNA Splicing Factors and RNA-Directed DNA Methylation. Biology 2014, 3, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. GENEVESTIGATOR V3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008. [Google Scholar] [CrossRef]
- Yoshida, S.; Foorno, D.; Cock, J.; Gomez, K. Laboratory Manual for Physiological Studies of Rice, 3rd ed.; International Rice Research Institute: Manila, Philippines, 1976. [Google Scholar]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Lohse, M.; Bolger, A.M.; Nagel, A.; Fernie, A.R.; Lunn, J.E.; Stitt, M.; Usadel, B. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012, 40, 622–627. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Chavez, L.; Jozefczuk, J.; Grimm, C.; Dietrich, J.; Timmermann, B.; Lehrach, H.; Herwig, R.; Adjaye, J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010, 20, 1441–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruntman, E.; Qi, Y.; Slotkin, R.K.; Roeder, T.; Martienssen, R.A.; Sachidanandam, R. Kismeth: Analyzer of plant methylation states through bisulfite sequencing. BMC Bioinform. 2008, 11, 371. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Condition | Biological Replicates | Total Reads | # Uniquely Mapped Reads | % Uniquely Mapped Reads | Cytosine Coverage % (Total C’s = 63095915) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0× | 1× | 2× | 3× | 4× | 5× | >5× | |||||
| Control 1 | 2 | 17.225.011 | 4.283.278 | 24.87 | 82.82 | 8.05 | 2.48 | 1.37 | 0.92 | 0.67 | 3.7 |
| Control 2 | 2 | 16.081.432 | 4.075.168 | 25.34 | 84.54 | 6.63 | 2.32 | 1.34 | 0.91 | 0.66 | 3.59 |
| Stress 1 | 2 | 13.681.641 | 3.639.466 | 26.60 | 82.86 | 8.89 | 2.46 | 1.29 | 0.84 | 0.61 | 3.05 |
| Stress 2 | 2 | 13.845.643 | 3.562.794 | 25.73 | 84.54 | 6.63 | 2.32 | 1.34 | 0.91 | 0.66 | 3.59 |
| Input | 1 | 14.661.478 | 7.016.939 | 47.86 | 51.2 | 22.17 | 14.59 | 7.39 | 3.03 | 1.06 | 0.57 |
| Chr | DMR ID | Coordinat. Start End | Repeat Masker Annotation | Gene Annotation | DMR Position Relative to the Gene | Gene Description |
|---|---|---|---|---|---|---|
I  | 1 | 3431001 3431100 | AnacC1 transposon (ORSiTETNOOT00122) | LOC_Os01g07270 | 78 bp downstream | Transposon |
| LOC_Os01g07280 | 506 bp downstream | Disease-resistance protein | ||||
| 2 | 38016401 38016700 | - | LOC_Os01g65490 | 2100 bp upstream | DNA binding protein | |
| LOC_Os01g65500 | 750 bp upstream | Chloride channel protein | ||||
| 3 | 39466301 39466500 | (CGG)n rich area | LOC_Os01g67910 | 5’ overlap | Expressed protein | |
| LOC_Os01g67920 | 796 bp downstream | Tetratricopeptide repeat protein | ||||
II  | 4 | 26500001 26500100 | - | LOC_Os02g43890 | Within (intron/exon/intron) | Hypothetical protein |
III | 5 | 36070201 36071200 | AnacA2 transposon (ORSiTETNOOT00130) | LOC_Os03g63840 | 4194 bp downstream | Expressed protein |
| LOC_Os03g63850 | 1972 bp upstream | OsFBDUF19 protein | ||||
IV  | 6 | 22831201 22831400 | (CGG)n rich area | LOC_Os04g38390 | 780 bp downstream | Wound/stress protein |
| LOC_Os04g38400 | 2620 bp upstream | Ethylene-insensitive 3 protein | ||||
V  | 7 | 4804401 4804700 | AnacA10 transposon (ORSiTETNOOT00124) | LOC_Os05g08760 | Within (exon/intron) | Expressed protein |
| 8 | 4805301 4805500 | - | LOC_Os05g08760 | Within (exon) | Expressed protein | |
| 9 | 9320201 9320400 | RIRE3 gypsy-type retrotransposon (ORSiTERTOOT00027) | LOC_Os05g16420 | 1570 bp downstream | SHR5-receptor-like kinase protein | |
| LOC_Os05g16430 | 1300 bp upstream | SHR5-receptor-like kinase protein | ||||
VI | 10 | 962901 963200 | E4 repeat sequence (ORSiOTOT00000050) | LOC_Os06g02680 | 680 bp upstream | Expressed protein |
| LOC_Os06g02690 | 20 bp downstream | Expressed protein | ||||
| 11 | 970501 970600 | - | LOC_Os06g02700 | Within (exon) | Retrotransposon Ty3-gypsy | |
| 12 | 983401 983500 | - | LOC_Os06g02730 | 3591 bp upstream | Aspartic proteinase nepenthesin-2 precursor protein | |
| LOC_Os06g02740 | 7261 bp upstream | Retrotransposon | ||||
| 13 | 1010401 1010700 | (CGG)n rich area | LOC_Os06g02770 | Within (exon) | Expressed gene | |
VIII | 14 | 9021501 9021600 | - | LOC_Os08g14950 | 1150 bp downstream | Receptor-like kinase 2 precursor protein |
| LOC_Os08g14960 | 4240 bp upstream | Receptor-like kinase precursor protein | ||||
IX  | 15 | 9475001 9475300 | Ty3-gypsy retrotransposon (ORSiTERT00200079) | LOC_Os09g15470 | 3500 bp upstream | Retrotransposon Ty3-gypsy |
| LOC_Os09g15480 | 1100bp downstream | Ser/Thr-rich protein | ||||
XI  | 16 | 20435601 20436000 | - | LOC_Os11g34870 | Within (intron) | Expressed protein |
XII  | 17 | 1446901 1447100 | AnacA10 transposon (ORSiTETNOOT00124) | LOC_Os12g03601 | 519 bp upstream | Expressed protein |
| LOC_Os12g03610 | 2283 bp upstream | Expressed protein | ||||
| 18 | 4989301 4989600 | noaCRR2 retrotransposon (ORSiTERTOOT00141) | LOC_Os12g09500 | 975 bp upstream | Cytochrome P450 protein | |
| LOC_Os12g09510 | 8570 bp upstream | Hypothetical protein | ||||
| 19 | 5108601 5108800 | Ty3-gypsy retrotransposon (ORSiTERT00200079) | LOC_Os12g09680 | Within (intron) | Retrotransposon Ty3-gypsy | |
| 20 | 5301501 5301700 | Centromere-like LTR transposon (ORSiCMCM00100011) | LOC_Os12g10000 | 2500 bp upstream | Retrotransposon | |
| LOC_Os12g10010 | 34 bp downstream | Expressed protein | ||||
| 21 | 25340601 25341000 | (GGA)n rich area | LOC_Os12g40930 | 155 bp upstream | Expressed protein | |
| LOC_Os12g40940 | 4377 bp upstream | Expressed protein | ||||
| 22 | 25763601 2764100 | noaCRR2 retrotransposon (ORSiTERTOOT00141) | LOC_Os12g41630 | 4000 bp upstream | OsFBX463–F-box domain protein | |
| LOC_Os12g41634 | Within (exon) | Expressed protein | ||||
| LOC_Os12g41640 | 800 bp upstream | Expressed protein |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, L.J.; Donoghue, M.T.A.; Barros, P.; Saibo, N.J.; Santos, A.P.; Oliveira, M.M. Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes 2019, 3, 4. https://doi.org/10.3390/epigenomes3010004
Ferreira LJ, Donoghue MTA, Barros P, Saibo NJ, Santos AP, Oliveira MM. Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes. 2019; 3(1):4. https://doi.org/10.3390/epigenomes3010004
Chicago/Turabian StyleFerreira, Liliana J., Mark T. A. Donoghue, Pedro Barros, Nelson J. Saibo, Ana Paula Santos, and M. Margarida Oliveira. 2019. "Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance" Epigenomes 3, no. 1: 4. https://doi.org/10.3390/epigenomes3010004
APA StyleFerreira, L. J., Donoghue, M. T. A., Barros, P., Saibo, N. J., Santos, A. P., & Oliveira, M. M. (2019). Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes, 3(1), 4. https://doi.org/10.3390/epigenomes3010004