
Epigenetic Signatures of Centrosomes Are Novel Targets in Cancer Diagnosis: Insights from an Analysis of the Cancer Genome Atlas


Abstract
:1. Introduction
2. Results
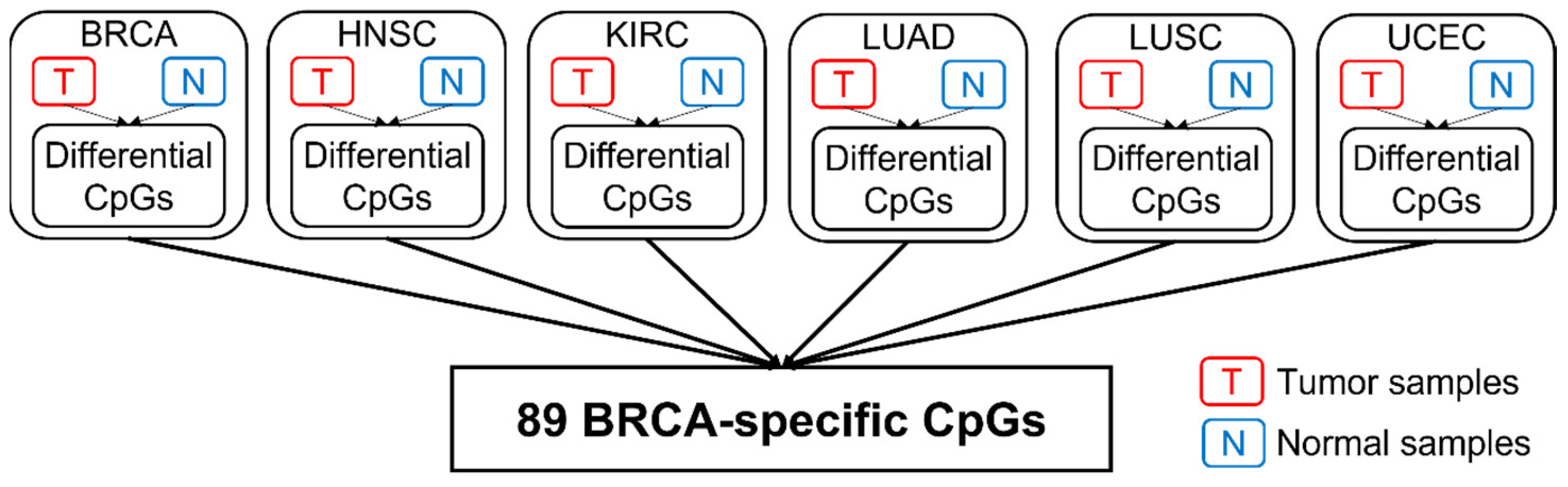
2.1. Identification of Cancer-Specific CpGs
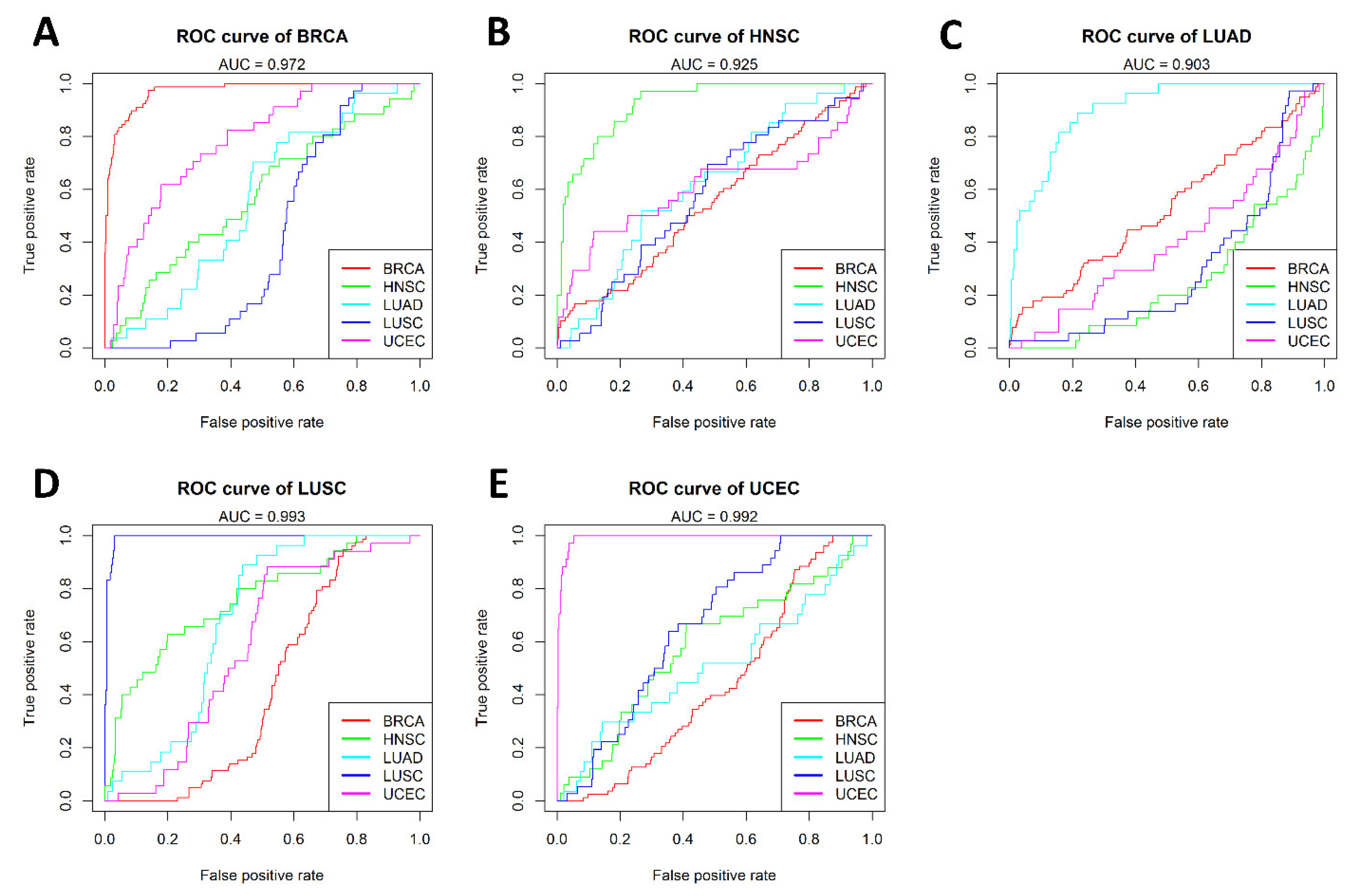
2.2. Cancer-Specific Epigenetic Model
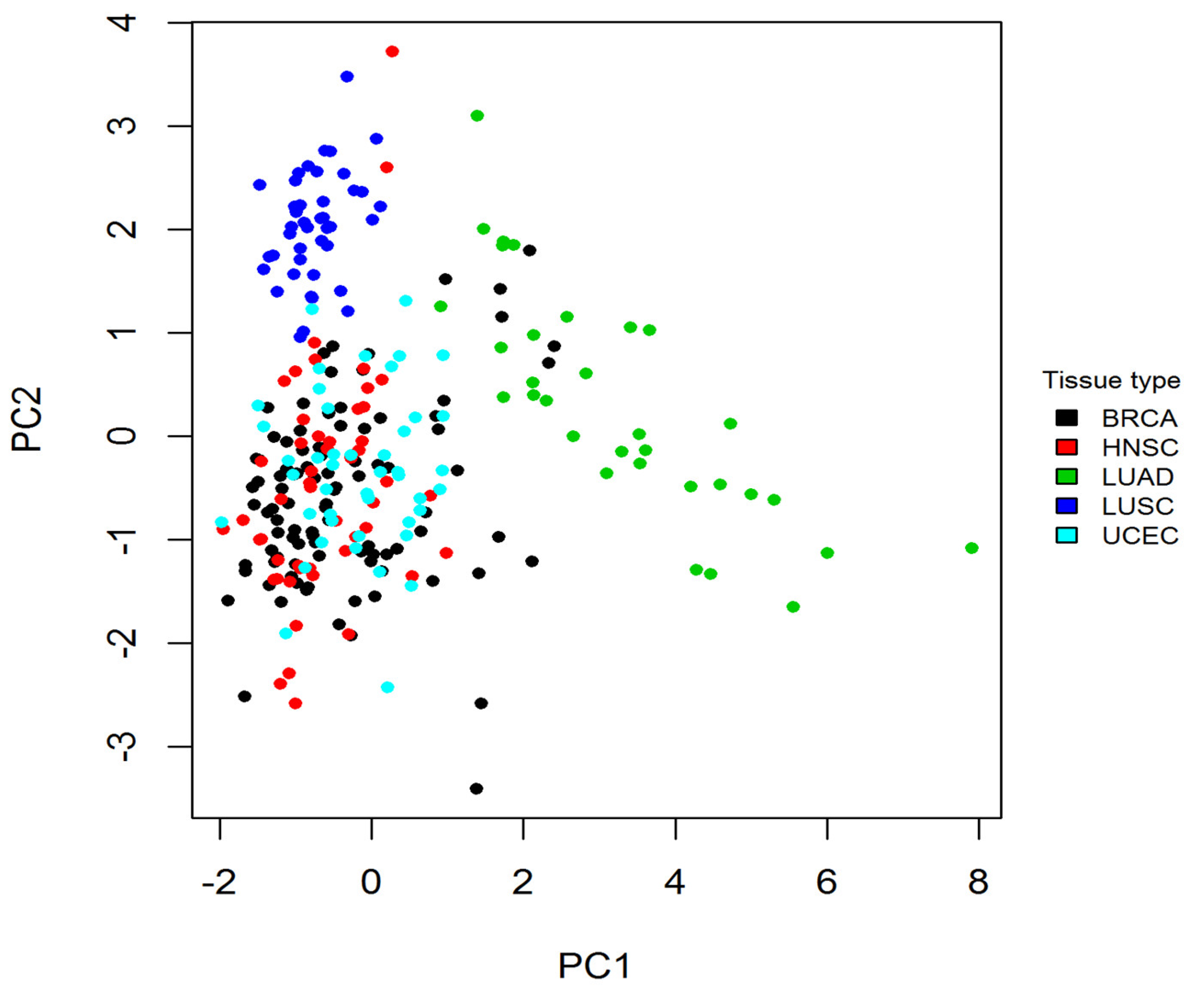
2.3. Tissue Specificity Underlying Cancer Type Specificity
2.4. Association Analysis between CpG and Gene Expression
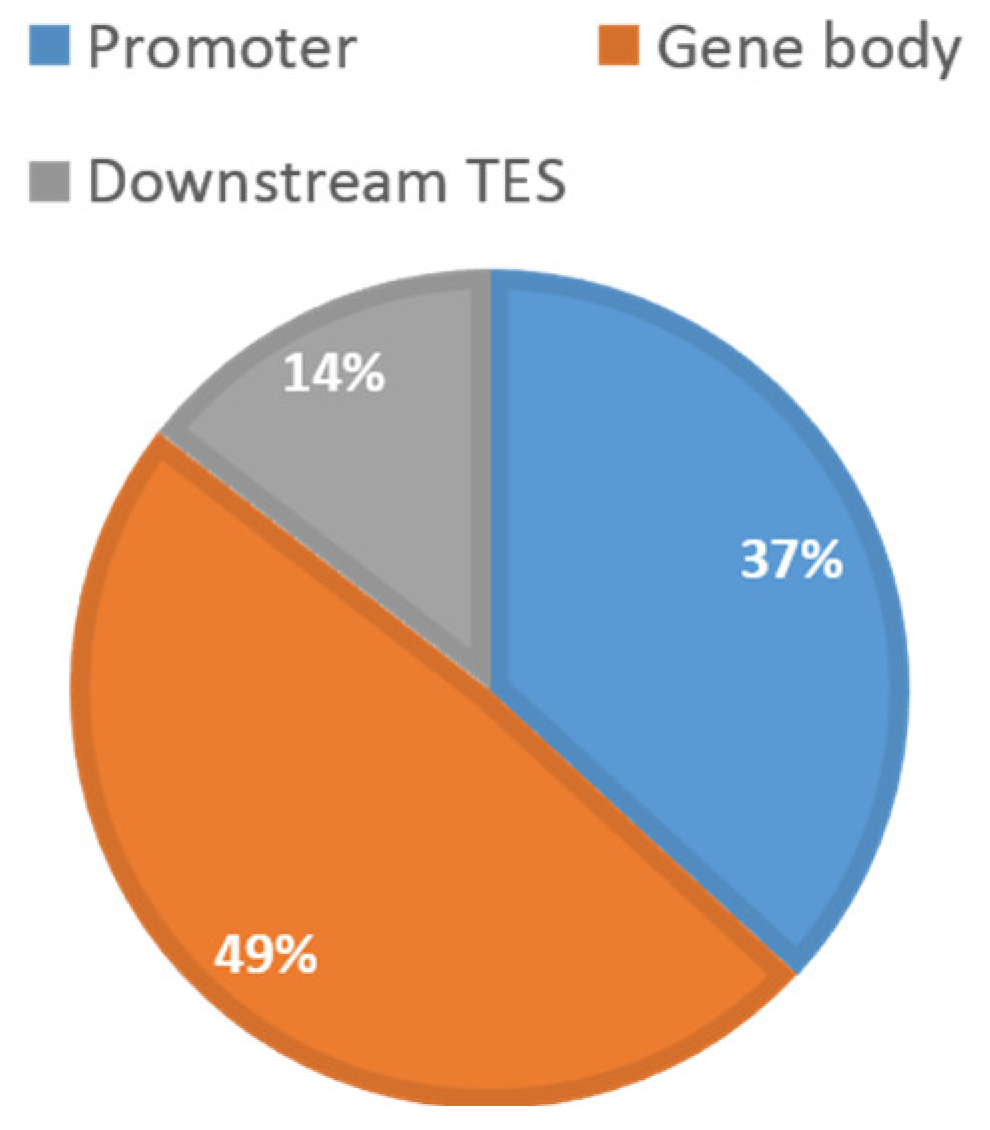
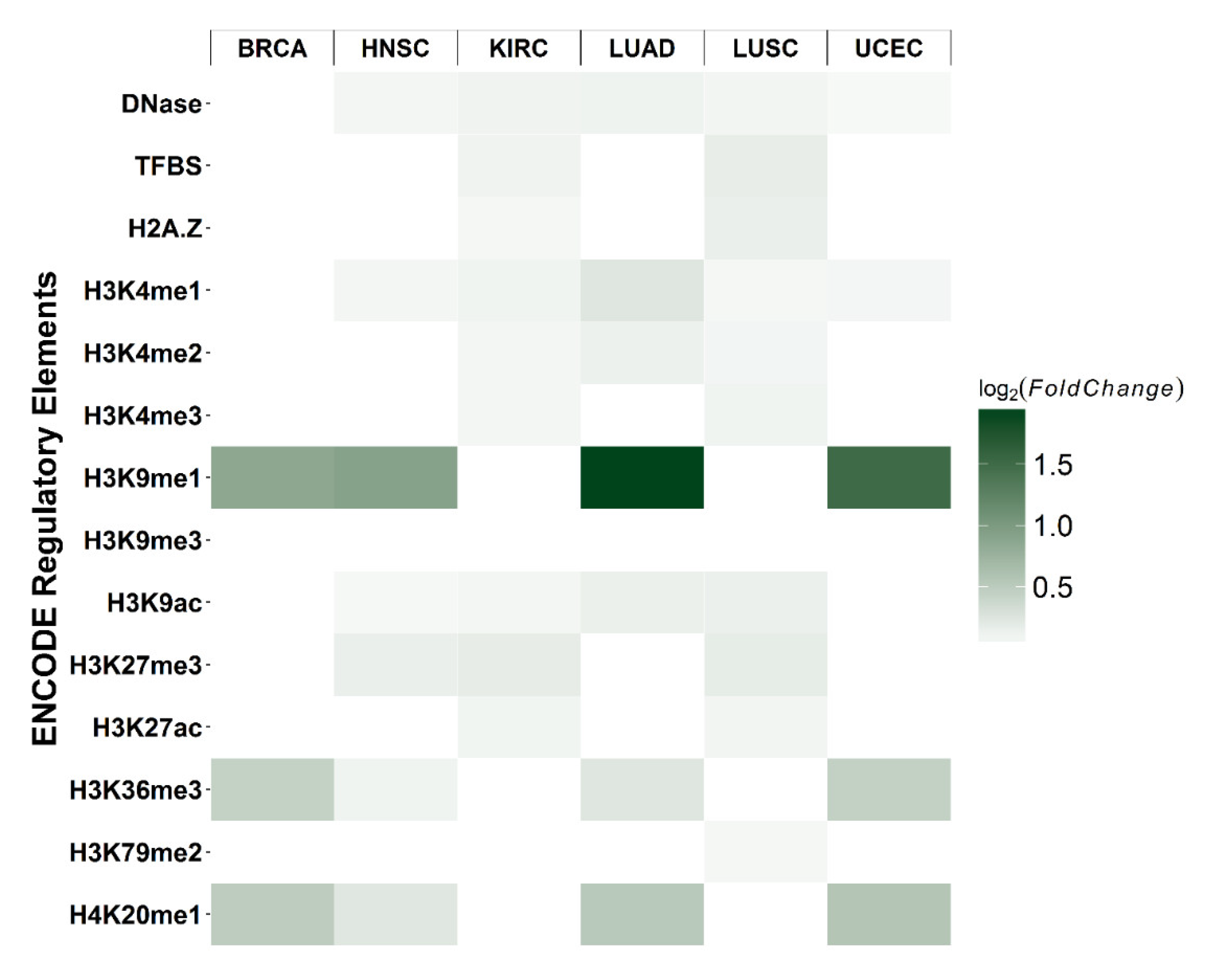
2.5. Co-Localization of Differential CpGs with Cis-Regulatory Elements
3. Discussion
4. Materials and Methods
4.1. TCGA Cancer Types
4.2. TCGA 450K Array Data
4.3. TCGA RNA-seq Data
4.4. Identify Cancer-Specific CpGs
4.5. Development of a Cancer-Specific Epigenetic Model
4.6. Linking DNA Methylation and Gene Expression
4.7. Co-localization of Differential CpGs with Cis-Regulatory Elements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Azimzadeh, J.; Bornens, M. Structure and duplication of the centrosome. J. Cell Sci. 2007, 120, 2139–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornens, M. The Centrosome in Cells and Organisms. Science 2012, 335, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M.; Gonczy, P. Centrosomes back in the limelight. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130452. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.; Wilkinson, C.; Mayor, T.; Mortensen, P.; Nigg, E.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, L.; Vanselow, K.; Skogs, M.; Toyoda, Y.; Lundberg, E.; Poser, I.; Falkenby, L.G.; Bennetzen, M.; Westendorf, J.; Nigg, E.; et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011, 30, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Glover, D.M. Structured illumination of the interface between centriole and peri-centriolar material. Open Biol. 2012, 2, 120104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennella, V.; Keszthelyi, B.; McDonald, K.L.; Chhun, B.; Kan, F.; Rogers, G.C.; Huang, B.; Agard, D. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat. Cell Biol. 2012, 14, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Dynlacht, B.D. Assembling a primary cilium. Curr. Opin. Cell Biol. 2013, 25, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonczy, P. Centrosomes and cancer: Revisiting a long-standing relationship. Nat. Rev. Cancer 2015, 15, 639–652. [Google Scholar] [CrossRef]
- Koutsami, M.; Tsantoulis, P.; Kouloukoussa, M.; Apostolopoulou, K.; Pateras, I.; Spartinou, Z.; Drougou, A.; Evangelou, K.; Kittas, C.; Bartkova, J.; et al. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J. Pathol. 2006, 209, 512–521. [Google Scholar] [CrossRef]
- Lingle, W.L.; Lutz, W.H.; Ingle, J.N.; Maihle, N.J.; Salisbury, J.L. Centrosome hypertrophy in human breast tumors: Implications for genomic stability and cell polarity. Proc. Natl. Acad. Sci. USA 1998, 95, 2950–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihan, G.A.; Purohit, A.; Wallace, J.; Knecht, H.; Woda, B.; Quesenberry, P.; Doxsey, S.J. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998, 58, 3974–3985. [Google Scholar] [PubMed]
- Hsu, L.C.; Kapali, M.; DeLoia, J.A.; Gallion, H.H. Centrosome abnormalities in ovarian cancer. Int. J. Cancer 2004, 113, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Mizumoto, K.; Nakamura, M.; Nakamura, K.; Kusumoto, M.; Niiyama, H.; Ogawa, T.; Tanaka, M. Centrosome abnormalities in pancreatic ductal carcinoma. Clin. Cancer Res. 1999, 5, 963–970. [Google Scholar]
- Krämer, A.; Neben, K.; Ho, A.D. Centrosome aberrations in hematological malignancies. Cell Biol. Int. 2005, 29, 375–383. [Google Scholar] [CrossRef]
- Giehl, M.; Fabarius, A.; Frank, O.; Hochhaus, A.; Häfner, M.; Hehlmann, R.; Seifarth, W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005, 19, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Esteller, M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Veenstra, G.J.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Kirkham, M.; Müller-Reichert, T.; Oegema, K.; Grill, S.; Hyman, A.A. SAS-4 Is a C. elegans Centriolar Protein that Controls Centrosome Size. Cell 2003, 112, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Kohlmaier, G.; Lončarek, J.; Meng, X.; McEwen, B.F.; Mogensen, M.M.; Spektor, A.; Dynlacht, B.D.; Khodjakov, A.; Gönczy, P. Overly Long Centrioles and Defective Cell Division upon Excess of the SAS-4-Related Protein CPAP. Curr. Biol. 2009, 19, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Cortessis, V.K.; Thomas, D.C.; Levine, A.J.; Breton, C.V.; Mack, T.M.; Siegmund, K.D.; Haile, R.W.; Laird, P.W. Environmental epigenetics: Prospects for studying epigenetic mediation of exposure–response relationships. Qual. Life Res. 2012, 131, 1565–1589. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Zhang, Z.; Wang, J.; Chiu, B.C.-H.; Hou, L.; Zhang, W. Application of the High-Throughput TAB-Array for the Discovery of Novel 5-Hydroxymethylcytosine Biomarkers in Pancreatic Ductal Adenocarcinoma. Epigenomes 2019, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, L.; Zhang, Z.; Zhang, X.; Lu, X.; Liu, W.; Shi, G.; Ge, Y.; Gao, P.; Yang, Y.; et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut 2019, 68, 2195–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altemose, N.; Logsdon, G.A.; Bzikadze, A.V.; Sidhwani, P.; Langley, S.A.; Caldas, G.V.; Hoyt, S.J.; Uralsky, L.; Ryabov, F.D.; Shew, C.J.; et al. Complete genomic and epigenetic maps of human centromeres. Science 2022, 376, eabl4178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mu, W.; Zhang, W. On the Analysis of the Illumina 450k Array Data: Probes Ambiguously Mapped to the Human Genome. Front. Genet. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071); TU Wien. 2017. Available online: https://CRAN.R-project.org/package=e1071 (accessed on 27 April 2022).
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | # Tumor Samples | # Normal Samples | # Tumor Samples | # Normal Samples | Cancer Type |
|---|---|---|---|---|---|
| 450K Array | RNA-seq | ||||
| BLCA | 418 | 21 | Bladder urothelial carcinomas | ||
| BRCA | 792 | 97 | 781 | 84 | Breast invasive carcinomas |
| COAD | 312 | 28 | Colon adenocarcinomas | ||
| GBM | 140 | 2 | Glioblastoma multiformes | ||
| HNSC | 528 | 50 | 520 | 20 | Head and neck squamous cell carcinomas |
| KIRC | 324 | 160 | 318 | 24 | Kidney renal papillary cell carcinomas |
| LUAD | 473 | 32 | 454 | 21 | Lung adenocarcinomas |
| LUSC | 370 | 42 | 370 | 8 | Lung squamous cell carcinomas |
| PAAD | 184 | 10 | Pancreatic adenocarcinomas | ||
| READ | 98 | 7 | Rectum adenocarcinomas | ||
| UCEC | 438 | 46 | 172 | 24 | Uterine corpus endometrial carcinomas |
| Total | 4077 | 505 | 2615 | 181 | |
| # Differential CpGs (FDR < 0.05) | # Hosting Genes a | # Cancer-Specific CpGs | # Hosting Genes b | Cancer-Specific Genes | |
|---|---|---|---|---|---|
| BRCA | 701 | 123 | 89 | 54 | CETN2, MAP7D3 |
| HNSC | 531 | 121 | 86 | 62 | - |
| KIRC | 1283 | 133 | 186 | 88 | PRKAR2B, CEP290, NOG, CDK5RAP2, HAUS6, PRKACB, PRKAR2A, NPHP4 |
| LUAD | 228 | 87 | 9 | 5 | - |
| LUSC | 1366 | 136 | 320 | 110 | RTTN, DCTN5, CEP120, IRAK1BP1, SSNA1, CEP135, ACTR1A, PCM1 |
| UCEC | 489 | 112 | 45 | 37 | ODF2, DCTN3, PIBF1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, W. Epigenetic Signatures of Centrosomes Are Novel Targets in Cancer Diagnosis: Insights from an Analysis of the Cancer Genome Atlas. Epigenomes 2022, 6, 14. https://doi.org/10.3390/epigenomes6020014
Zhang Z, Zhang W. Epigenetic Signatures of Centrosomes Are Novel Targets in Cancer Diagnosis: Insights from an Analysis of the Cancer Genome Atlas. Epigenomes. 2022; 6(2):14. https://doi.org/10.3390/epigenomes6020014
Chicago/Turabian StyleZhang, Zhou, and Wei Zhang. 2022. "Epigenetic Signatures of Centrosomes Are Novel Targets in Cancer Diagnosis: Insights from an Analysis of the Cancer Genome Atlas" Epigenomes 6, no. 2: 14. https://doi.org/10.3390/epigenomes6020014
APA StyleZhang, Z., & Zhang, W. (2022). Epigenetic Signatures of Centrosomes Are Novel Targets in Cancer Diagnosis: Insights from an Analysis of the Cancer Genome Atlas. Epigenomes, 6(2), 14. https://doi.org/10.3390/epigenomes6020014