
Epigenetic Regulation in Breast Cancer: Insights on Epidrugs


Abstract
:1. Introduction
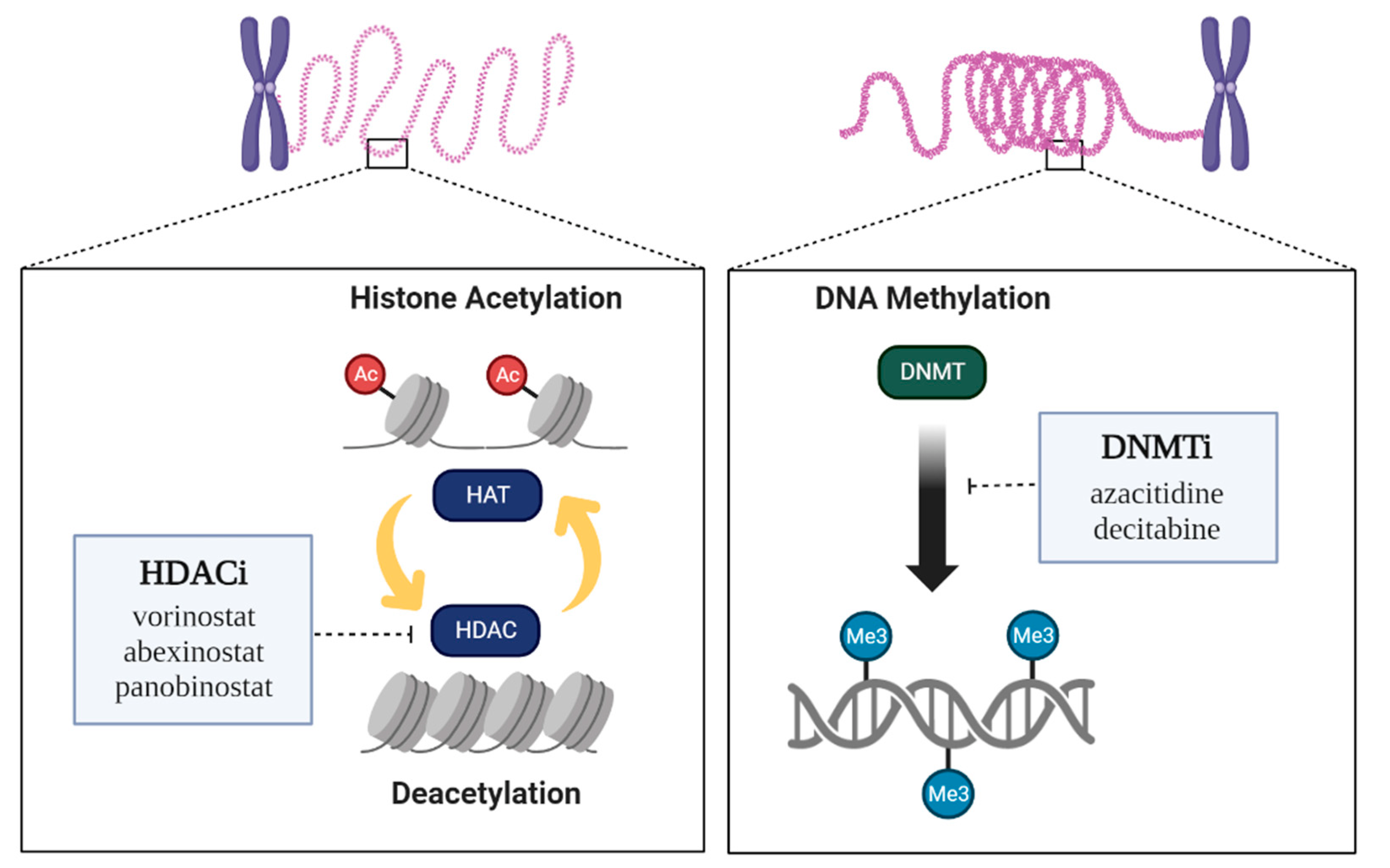
2. Regulatory Enzymes of Histone Methylation and Acetylation
3. Epigenetic Regulation in Breast Cancer
3.1. Role of Noncoding RNA in Epigenetic Regulation
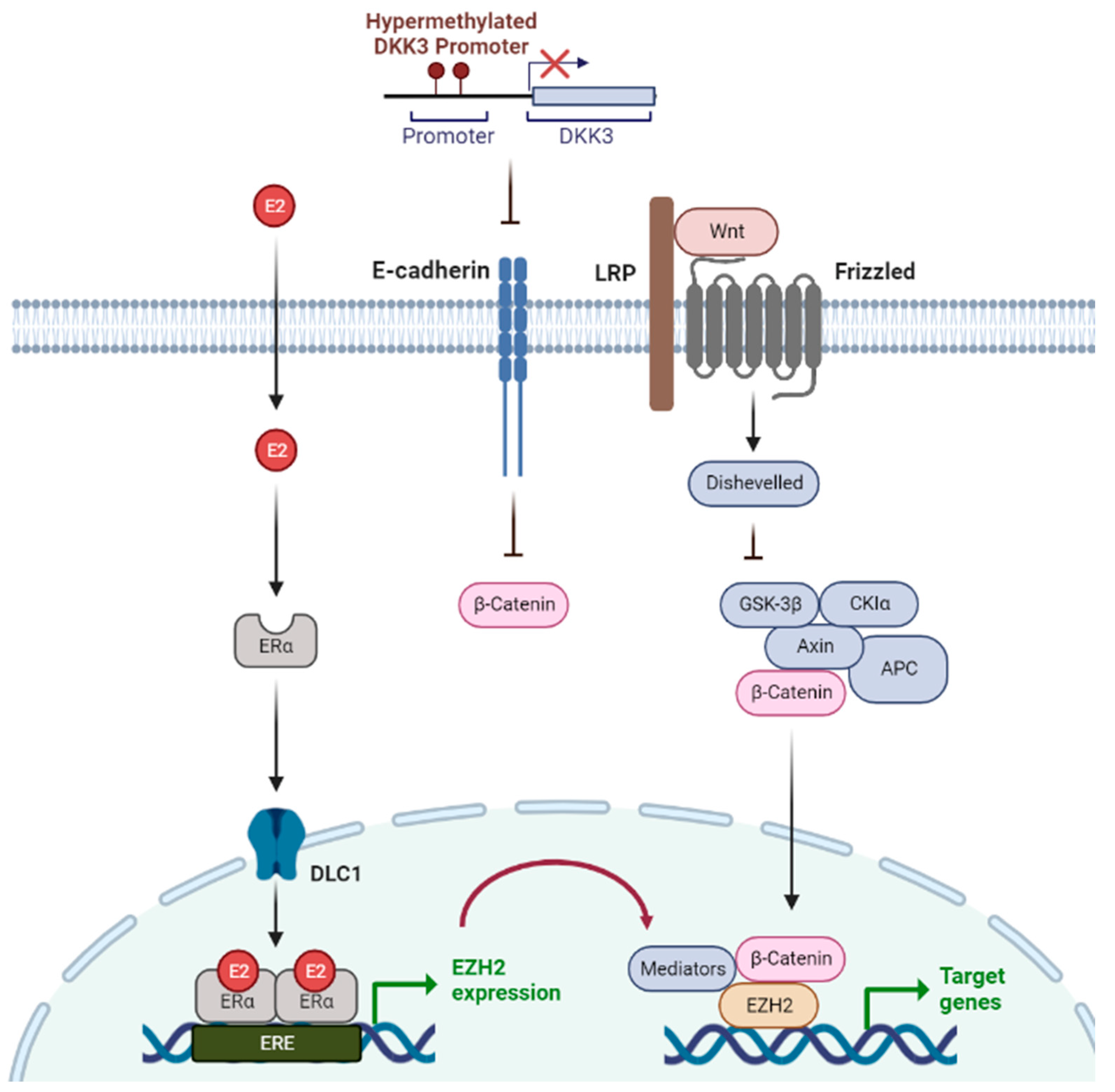
3.2. Estrogen-Related Epigenetic Mechanisms
3.3. Epigenetic Modulation during Epithelial to Mesenchymal Transition (EMT) in Breast Cancer
4. Epidrug
4.1. DNMT Inhibitors (DNMTis)
4.2. HDAC Inhibitors
4.3. Recently Developed Epidrugs
5. Epidrugs in Breast Cancer
5.1. DNMTi in Breast Cancer
5.2. HDACi in Breast Cancer
5.3. Combination Therapy with Epidrugs in Breast Cancer
5.4. Epidrugs with Nanotechnology
6. Trends in Clinical Trials of Epidrugs in Breast Cancer
6.1. Clinical Trials in Breast Cancer
6.2. Limitations and Prospects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA: A Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Lu, H.-M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51. [Google Scholar] [CrossRef]
- Wooster, R.; Weber, B.L. Breast and Ovarian Cancer. New Engl. J. Med. 2003, 348, 2339–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, A.C.; Spurdle, A.B.; Sinilnikova, O.M.; Healey, S.; Pooley, K.A.; Schmutzler, R.K.; Versmold, B.; Engel, C.; Meindl, A.; Arnold, N.; et al. Common Breast Cancer-Predisposition Alleles Are Associated with Breast Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. Am. J. Hum. Genet. 2008, 82, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Pedroza, D.A.; Rajamanickam, V.; Subramani, R.; Bencomo, A.; Galvez, A.; Lakshmanaswamy, R. Progesterone receptor membrane component 1 promotes the growth of breast cancers by altering the phosphoproteome and augmenting EGFR/PI3K/AKT signalling. Br. J. Cancer 2020, 123, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Barahona, V.; Joshi, R.S.; Esteller, M. Use of DNA methylation profiling in translational oncology. Semin. Cancer Biol. 2022, 83, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Herek, T.A.; Bouska, A.; Lone, W.; Sharma, S.; Amador, C.; Heavican, T.B.; Li, Y.; Wei, Q.; Jochum, D.; Greiner, T.C.; et al. DNMT3A mutations define a unique biological and prognostic subgroup associated with cytotoxic T cells in PTCL-NOS. Blood 2022, 140, 1278–1290. [Google Scholar] [CrossRef]
- Loberg, M.A.; Bell, R.K.; Goodwin, L.O.; Eudy, E.; Miles, L.A.; SanMiguel, J.M.; Young, K.; Bergstrom, D.E.; Levine, R.L.; Schneider, R.K.; et al. Sequentially inducible mouse models reveal that Npm1 mutation causes malignant transformation of Dnmt3a-mutant clonal hematopoiesis. Leukemia 2019, 33, 1635–1649. [Google Scholar] [CrossRef] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: What's next? J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Banks, K.M.; Pan, H.; Verma, N.; Dixon, G.R.; Zhou, T.; Ding, B.; Elemento, O.; Chen, S.; Huangfu, D.; et al. Stage-specific regulation of DNA methylation by TET enzymes during human cardiac differentiation. Cell Rep. 2021, 37, 110095. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc'his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics 2019, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Jorge, A.L.; Pereira, E.R.; Oliveira, C.S.D.; Ferreira, E.D.S.; Menon, E.T.N.; Diniz, S.N.; Pezuk, J.A. MicroRNAs: Understanding their role in gene expression and cancer. Einstein 2021, 19, eRB5996. [Google Scholar] [CrossRef]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, U.; Faheem, M.; Qadri, I.; Begum, N.; Yassine, H.M.; Al Thani, A.A.; Mathew, S. Expression profile of MicroRNA: An Emerging Hallmark of Cancer. Curr. Pharm. Des. 2019, 25, 642–653. [Google Scholar] [CrossRef]
- Mondal, P.; Meeran, S.M. Long non-coding RNAs in breast cancer metastasis. Noncoding RNA Res. 2020, 5, 208–218. [Google Scholar] [CrossRef]
- Filippova, E.A.; Fridman, M.V.; Burdennyy, A.M.; Loginov, V.I.; Pronina, I.V.; Lukina, S.S.; Dmitriev, A.A.; Braga, E.A. Long Noncoding RNA GAS5 in Breast Cancer: Epigenetic Mechanisms and Biological Functions. Int. J. Mol. Sci. 2021, 22, 6810. [Google Scholar] [CrossRef]
- Thomas, M.P.; Potter, B.V. The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Biol. 2013, 137, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Fernandez, S.V.; Russo, P.A.; Fernbaugh, R.; Sheriff, F.S.; Lareef, H.M.; Garber, J.; Russo, I.H. 17-Beta-Estradiol induces transformation and tumorigenesis in human breast epithelial cells. FASEB J. 2006, 20, 1622–1634. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, M.D.; Shaheer, K. Endocrine disrupting chemicals may deregulate DNA repair through estrogen receptor mediated seizing of CBP/p300 acetylase. J. Endocrinol. Investig. 2020, 43, 1189–1196. [Google Scholar] [CrossRef]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Mir, R.; Galande, S. Epigenetic Regulation of the Wnt/beta-Catenin Signaling Pathway in Cancer. Front. Genet. 2021, 12, 681053. [Google Scholar] [CrossRef]
- Khan, Z.; Arafah, M.; Shaik, J.P.; Mahale, A.; Alanazi, M.S. High-frequency deregulated expression of Wnt signaling pathway members in breast carcinomas. Onco Targets Ther. 2018, 11, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Nowak, E.; Bednarek, I. Aspects of the Epigenetic Regulation of EMT Related to Cancer Metastasis. Cells 2021, 10, 3435. [Google Scholar] [CrossRef]
- Liu, J.; Sun, X.; Qin, S.; Wang, H.; Du, N.; Li, Y.; Pang, Y.; Wang, C.; Xu, C.; Ren, H. CDH1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncol. Lett. 2016, 11, 2635–2643. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, W.; Hao, M. Phenethyl isothiocyanate reduces breast cancer stem cell-like properties by epigenetic reactivation of CDH1. Oncol. Rep. 2020, 45, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; El Hachlafi, N.; Aanniz, T.; Bourais, I.; Mechchate, H.; Benali, T.; Shariati, M.A.; Burkov, P.; Lorenzo, J.M.; Wilairatana, P.; et al. Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates. Molecules 2022, 27, 2568. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yang, Y.; Wang, Y. Predictive biomarkers and potential drug combinations of epi-drugs in cancer therapy. Clin. Epigenetics 2021, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, T.; Luo, H.; Liu, Y.; Liu, D. Targeting Epigenetic Regulatory Enzymes for Cancer Therapeutics: Novel Small-Molecule Epidrug Development. Front. Oncol. 2022, 12, 848221. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Ramos, F. Pre-emptive azacitidine for relapse prevention in acute myeloid leukaemia. Lancet Oncol. 2018, 19, 1557–1558. [Google Scholar] [CrossRef]
- Montalvo-Casimiro, M.; Gonzalez-Barrios, R.; Meraz-Rodriguez, M.A.; Juarez-Gonzalez, V.T.; Arriaga-Canon, C.; Herrera, L.A. Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 2020, 10, 605386. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2010, 3, 166–179. [Google Scholar]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Bruserud, O.; Stapnes, C.; Ersvaer, E.; Gjertsen, B.T.; Ryningen, A. Histone deacetylase inhibitors in cancer treatment: A review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr. Pharm. Biotechnol. 2007, 8, 388–400. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, P.; Yin, X.; Zhang, J.; Huo, M.; Gao, J.; Li, G.; Teng, X.; Yu, H.; Huang, W.; et al. The histone deacetylase inhibitor PCI-24781 impairs calcium influx and inhibits proliferation and metastasis in breast cancer. Theranostics 2021, 11, 2058–2076. [Google Scholar] [CrossRef] [PubMed]
- Buggy, J.J.; Cao, Z.A.; Bass, K.E.; Verner, E.; Balasubramanian, S.; Liu, L.; Schultz, B.E.; Young, P.R.; Dalrymple, S.A. CRA-024781: A novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2006, 5, 1309–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. Epidrugs in the Immunotherapy of Cutaneous and Uveal Melanoma. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. -Anti-Cancer Agents) 2017, 17, 190–205. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Rothermundt, C.; Andreou, D.; Blay, J.Y.; Brodowicz, T.; Desar, I.M.E.; Dileo, P.; Gelderblom, H.; Haas, R.; Jakob, J.; Jones, R.L.; et al. Controversies in the management of patients with soft tissue sarcoma: Recommendations of the Conference on State of Science in Sarcoma 2022. Eur. J. Cancer 2023, 180, 158–179. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An emerging therapeutic modality in precision medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Paiva, S.-L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2019, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Richart, L.; Margueron, R. Drugging histone methyltransferases in cancer. Curr. Opin. Chem. Biol. 2020, 56, 51–62. [Google Scholar] [CrossRef]
- Dey, A.; Kundu, M.; Das, S.; Jena, B.C.; Mandal, M. Understanding the function and regulation of Sox2 for its therapeutic potential in breast cancer. Biochim. et Biophys. Acta (BBA)—Rev. Cancer 2022, 1877, 188692. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, C.; Yu, Z.; Li, X.; Mu, Q.; Liao, G.; Yu, B. Natural products as LSD1 inhibitors for cancer therapy. Acta Pharm. Sin. B 2021, 11, 621–631. [Google Scholar] [CrossRef]
- Cuyas, E.; Gumuzio, J.; Verdura, S.; Brunet, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Alarcon, T.; Encinar, J.A.; Martin, A.G.; Menendez, J.A. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: A potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging (Albany NY) 2020, 12, 4794–4814. [Google Scholar] [CrossRef]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef] [Green Version]
- Vernier, M.; McGuirk, S.; Dufour, C.R.; Wan, L.; Audet-Walsh, E.; St-Pierre, J.; Giguere, V. Inhibition of DNMT1 and ERRalpha crosstalk suppresses breast cancer via derepression of IRF4. Oncogene 2020, 39, 6406–6420. [Google Scholar] [CrossRef]
- Butler, C.; Sprowls, S.; Szalai, G.; Arsiwala, T.; Saralkar, P.; Straight, B.; Hatcher, S.; Tyree, E.; Yost, M.; Kohler, W.J.; et al. Hypomethylating Agent Azacitidine Is Effective in Treating Brain Metastasis Triple-Negative Breast Cancer Through Regulation of DNA Methylation of Keratin 18 Gene. Transl. Oncol. 2020, 13, 100775. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Gilmer, J.-J. Combination Therapies of Guadecitabine and Immune Checkpoint Inhibitors in a Murine Triple-Negative Breast Cancer Model. Virginia Commonwealth University Research Poster 2020. [Google Scholar]
- Ayipo, Y.O.; Ajiboye, A.T.; Osunniran, W.A.; Jimoh, A.A.; Mordi, M.N. Epigenetic oncogenesis, biomarkers and emerging chemotherapeutics for breast cancer. Biochim. et Biophys. Acta (BBA)—Gene Regul. Mech. 2022, 1865, 194873. [Google Scholar] [CrossRef]
- Chequin, A.; Costa, L.E.; de Campos, F.F.; Moncada, A.D.B.; de Lima, L.T.F.; Sledz, L.R.; Picheth, G.F.; Adami, E.R.; Acco, A.; Goncalves, M.B.; et al. Antitumoral activity of liraglutide, a new DNMT inhibitor in breast cancer cells in vitro and in vivo. Chem. Interactions 2021, 349, 109641. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z.; Wang, H.; Liu, X.; Zhou, Z.; Tang, J.; Liu, X.; Zheng, M.; Shen, Y. SAHA (vorinostat) facilitates functional polymer-based gene transfection via upregulation of ROS and synergizes with TRAIL gene delivery for cancer therapy. J. Drug Target. 2019, 27, 306–314. [Google Scholar] [CrossRef]
- Patra, S.; Praharaj, P.P.; Klionsky, D.J.; Bhutia, S.K. Vorinostat in autophagic cell death: A critical insight into autophagy-mediated, -associated and -dependent cell death for cancer prevention. Drug Discov. Today 2021, 27, 269–279. [Google Scholar] [CrossRef]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, S.; Shen, T.; Lu, H.; Xiao, D.; Zhao, M.; Yao, Y.; Li, X.; Zhang, G.; Zhou, X.; et al. Trichostatin A reverses epithelial-mesenchymal transition and attenuates invasion and migration in MCF-7 breast cancer cells. Exp. Ther. Med. 2020, 19, 1687–1694. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pan, X.; Li, Y.; Wang, R.; Yang, Y.; Jiang, B.; Sun, G.; Shao, C.; Wang, M.; Gong, Y. CUL4B renders breast cancer cells tamoxifen-resistant via miR-32-5p/ER-alpha36 axis. J. Pathol. 2021, 254, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Li, Y.; Xu, X.; Wang, X.; Zhang, K.; Tang, Y.; Qiu, H.; Shi, D.; Zhang, C.; Long, Q.; et al. Panobinostat (LBH589) inhibits Wnt/beta-catenin signaling pathway via upregulating APCL expression in breast cancer. Cell. Signal. 2019, 59, 62–75. [Google Scholar] [CrossRef]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J. Immunother. Cancer 2020, 8, e000195. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zhang, J.; Yan, C.; Li, X.; Zhang, J.; Ling, R. Small molecule HDAC inhibitors: Promising agents for breast cancer treatment. Bioorganic Chem. 2019, 91, 103184. [Google Scholar] [CrossRef]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Tsai, Y.-C.; Tsai, T.-H.; Kuo, K.-L.; Su, Y.-F.; Chang, C.-H.; Lin, C.-L. Valproic acid reduces vasospasm through modulation of akt phosphorylation and attenuates neuronal apoptosis in subarachnoid hemorrhage rats. Int. J. Mol. Sci. 2021, 22, 5975. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov. Today 2019, 24, 685–702. [Google Scholar] [CrossRef]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Investig. Dermatol. 2021, 141, 903–912. [Google Scholar] [CrossRef]
- Exman, P.; Barroso-Sousa, R.; Tolaney, S.M. Evidence to date: Talazoparib in the treatment of breast cancer. Onco Targets Ther. 2019, 12, 5177–5187. [Google Scholar] [CrossRef] [Green Version]
- Buocikova, V.; Rios-Mondragon, I.; Pilalis, E.; Chatziioannou, A.; Miklikova, S.; Mego, M.; Pajuste, K.; Rucins, M.; Yamani, N.E.; Longhin, E.M.; et al. Epigenetics in Breast Cancer Therapy-New Strategies and Future Nanomedicine Perspectives. Cancers 2020, 12, 3622. [Google Scholar] [CrossRef]
- Yellapu, N.K.; Ly, T.; Sardiu, M.E.; Pei, D.; Welch, D.R.; Thompson, J.A.; Koestler, D.C. Synergistic anti-proliferative activity of JQ1 and GSK2801 in triple-negative breast cancer. BMC Cancer 2022, 22, 627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, L.; Ge, G.; Hu, K. Emerging Epigenetic-Based Nanotechnology for Cancer Therapy: Modulating the Tumor Microenvironment. Adv. Sci. 2023, 1, e2206169. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.; Liu, Y.; Xin, D.; Lei, W.; Liang, A.; Han, W.; Qian, W. Epi-immunotherapy for cancers: Rationales of epi-drugs in combination with immunotherapy and advances in clinical trials. Cancer Commun. 2022, 42, 493–516. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Gueugnon, F.; Boutin, B.; Guillot, F.; Blanquart, C.; Rogel, A.; Padieu, M.; Pouliquen, D.; Fonteneau, J.F.; Gregoire, M. A 5-aza-2'-deoxycytidine/valproate combination induces cytotoxic T-cell response against mesothelioma. Eur. Respir. J. 2011, 38, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.S.; Lee, Z.Y.; Chuah, L.H.; Mai, C.W.; Ngai, S.C. Epigenetics in Metastatic Breast Cancer: Its Regulation and Implications in Diagnosis, Prognosis and Therapeutics. Curr. Cancer Drug Targets 2019, 19, 82–100. [Google Scholar] [CrossRef]
- Cai, F.F.; Kohler, C.; Zhang, B.; Wang, M.H.; Chen, W.J.; Zhong, X.Y. Epigenetic therapy for breast cancer. Int. J. Mol. Sci. 2011, 12, 4465–4487. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- de Lera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitargeted drugs. Clin. Epigenetics 2016, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hao, D.; Wang, L.; Wang, H.; Wang, Y.; Zhao, Z.; Li, P.; Deng, C.; Di, L.J. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy. Sci. Rep. 2017, 7, 4035. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.; Castellano, J.M.; Onuma, O.K. Putting polypills into practice: Challenges and lessons learned. Lancet 2017, 389, 1066–1074. [Google Scholar] [CrossRef]
- Benedetti, R.; Conte, M.; Iside, C.; Altucci, L. Epigenetic-based therapy: From single- to multi-target approaches. Int. J. Biochem. Cell. Biol. 2015, 69, 121–131. [Google Scholar] [CrossRef]
- Daher-Reyes, G.S.; Merchan, B.M.; Yee, K.W.L. Guadecitabine (SGI-110): An investigational drug for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Expert. Opin. Investig. Drugs 2019, 28, 835–849. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subtype (Class) | Epidrug Name | Effect | Combination | FDA-Approval | Reference |
|---|---|---|---|---|---|
| DNA methyltransferase inhibitor (DNMTi) | 5-azacitidine | Treat myelodysplastic syndrome | Vaprobic acid Entinostat | Yes | [36] |
| Decitabine | Treat myelodysplastic syndrome and acute myeloid leukemia (AML) | Zebulin | Yes | [36] | |
| Guadecitabine | Treats myeloid malignancies and inhibits tumor growth | Pembrolizumab | Yes | [93] | |
| Isoflavones | Treats prostate cancer and inhibits thyroid hormone production. | Resveratrol | No | [12,33] | |
| Hydralazine | Treats high blood pressure and heart failure | Isosorbide dinitrate | Yes | [12,33] | |
| Histone deacetylase inhibitor (HDACi) | Vorinostat | Inhibits the proliferation of TNBC cell; Treat cutaneous T-cell lymphoma | PKF118-310 Simvastatin letrozole | Yes | [41,68] |
| Abexinostat | Treats non-cell cycle-specific cytotoxicity and 4-line follicular lymphoma. | Pembrolizumab | Yes | [43] | |
| Panobinostat | Treats multiple myeloma in combination with other drugs. | Trastuzumab Letrozole | Yes | [45,72] | |
| Trichostatin A | Inhibits invasive and migratory abilities of breast cancer cells | Cisplatin Gemcitabine Doxorubicin | No | [70] | |
| Lysine-specific demethylase 1 inhibitor (LSD1i) | Iadademstat | Acts as an immunomodulator and candidate for therapeutic combinations in leukemia or some solid tumors. | Azacitidine Venetoclax | Yes | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, A.; Mo, K.; Kwon, H.; Choe, S.; Park, M.; Kwak, W.; Yoon, H. Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes 2023, 7, 6. https://doi.org/10.3390/epigenomes7010006
Kim A, Mo K, Kwon H, Choe S, Park M, Kwak W, Yoon H. Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes. 2023; 7(1):6. https://doi.org/10.3390/epigenomes7010006
Chicago/Turabian StyleKim, Ayoung, Kyumin Mo, Hyeonseok Kwon, Soohyun Choe, Misung Park, Woori Kwak, and Hyunho Yoon. 2023. "Epigenetic Regulation in Breast Cancer: Insights on Epidrugs" Epigenomes 7, no. 1: 6. https://doi.org/10.3390/epigenomes7010006
APA StyleKim, A., Mo, K., Kwon, H., Choe, S., Park, M., Kwak, W., & Yoon, H. (2023). Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes, 7(1), 6. https://doi.org/10.3390/epigenomes7010006