DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Technical Details and Definitions
2.1. Computational Details
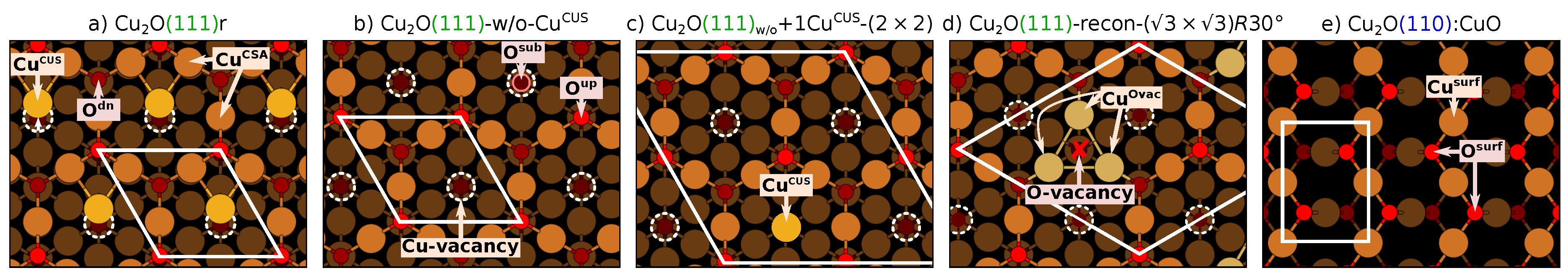
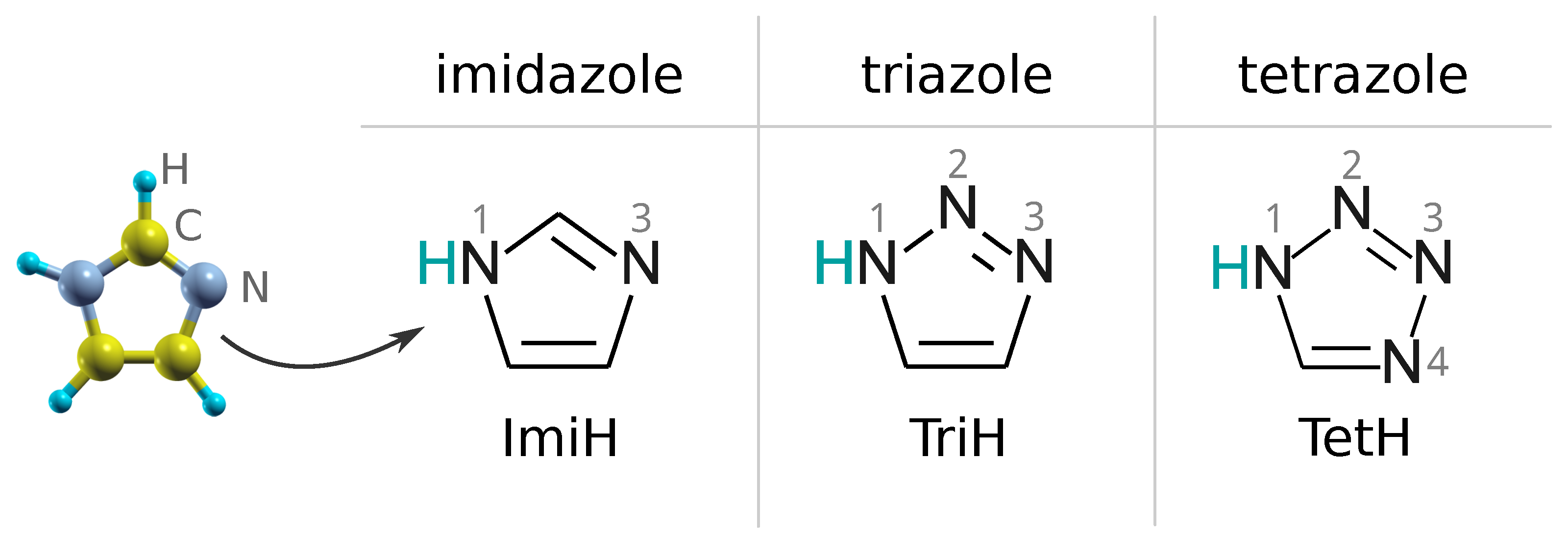
2.2. CuO as a Model of Oxidized Copper Surface and Description of Considered Surface Models
- (i)
- stoichiometric CuO(111)—this surface contains two distinct copper ions: a coordinatively saturated (CSA) and coordinatively unsaturated (CUS), labeled as Cu and Cu, respectively. It also contains two distinct oxygen ions: O and O located above (up) and below (dn) the surface Cu layer, respectively. Note that the high-symmetry CuO(111) is not stable and when the symmetry is broken the Cu ions relax laterally toward adjacent Cu ions [19]; the corresponding structure is designated as CuO(111)r, where “r” stands for “relaxed”.
- (ii)
- CuO(111)-w/o-Cu—this model is the CuO(111) that lacks all the Cu ions; notation “-w/o-Cu” stands for “CuO(111) without Cu ions”.
- (iii)
- CuO(111)+1Cu—this model is the CuO(111) that lacks all but a single Cu ion either per adsorbed molecule or per supercell; the subscript “w/o” is a shorthand for “w/o-Cu” and the suffix “+1Cu” conveys that one Cu ion is retained.
- (iv)
- CuO(111)-recon-( × )R30—this surface is derived from CuO(111)-w/o-Cu by removing one third of O ions such that the resulting oxygen vacancies form an ordered ( × )R30 pattern. Each O-vacancy is surrounded by three coordinatively unsaturated Cu ions, labeled as Cu. The Cu ions therefore always appear in triplets and we use the term “Cu site” to designate the site composed of these three ions.
- (v)
- CuO(110):CuO—this is the CuO(110) model terminated by a CuO layer on both sides of the slab. Note that the stoichiometric CuO(110) slab consists of CuO–Cu bilayers and is thus polar (terminated with a CuO layer on one side and a Cu layer on the other side). Hence, a symmetric slab terminated with the CuO layer on both sides is used instead; thus the denotation CuO(110):CuO. The surface CuO layer of CuO(110):CuO consists of coordinatively saturated Cu ions, labeled as Cu, and O ions labeled as O.
The Difference Between CuO(111)+1Cu and CuO(111)+1Cu–(N × N) Designations
2.3. Adsorption Calculations
2.3.1. Definition of Surface Coverage
2.3.2. Non-Dissociative, Dissociative, Mixed-Site, and Mixed-Mode Adsorption
2.3.3. Adsorption Equations
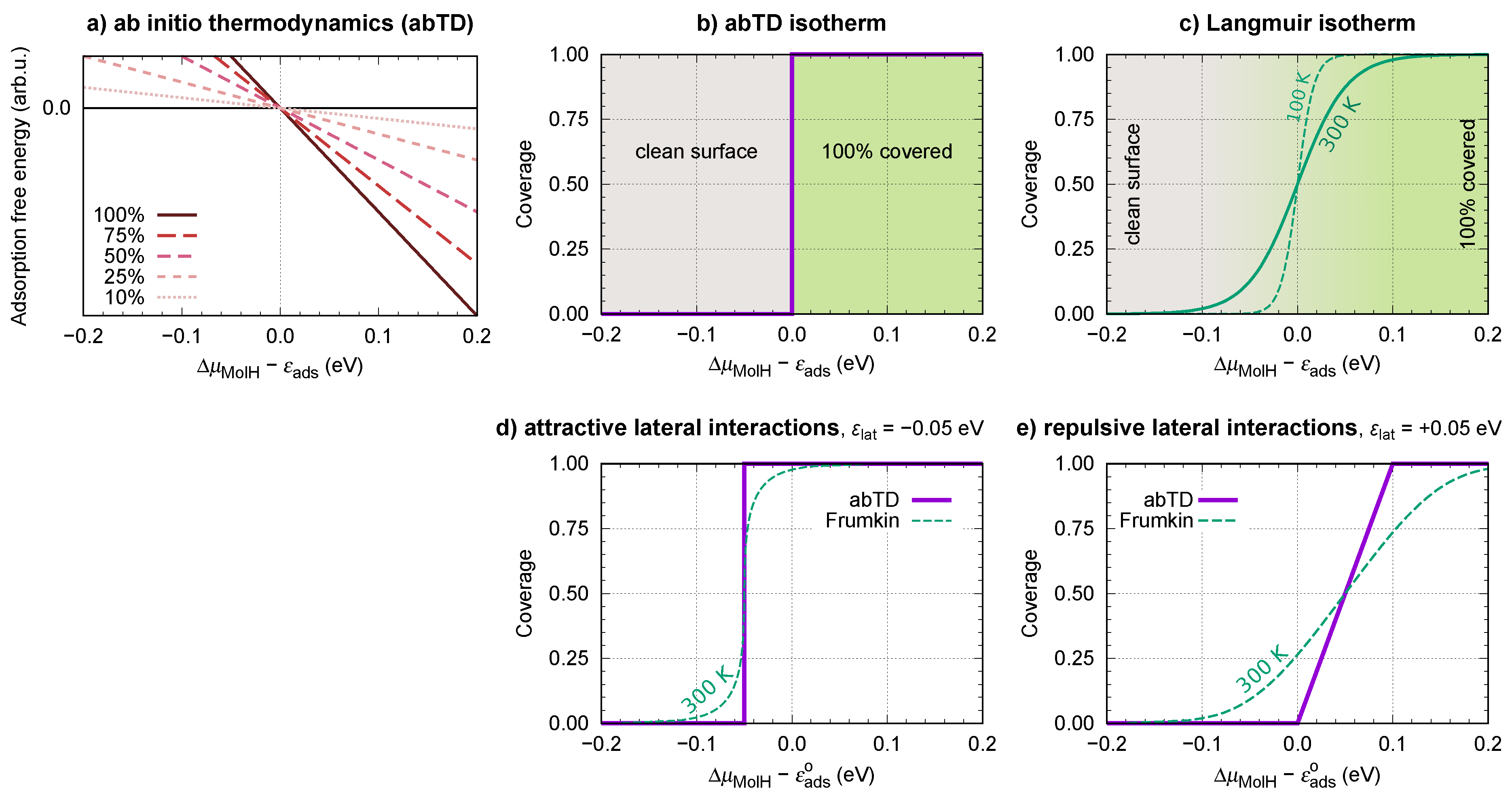
2.4. Thermodynamic Stability
3. Results
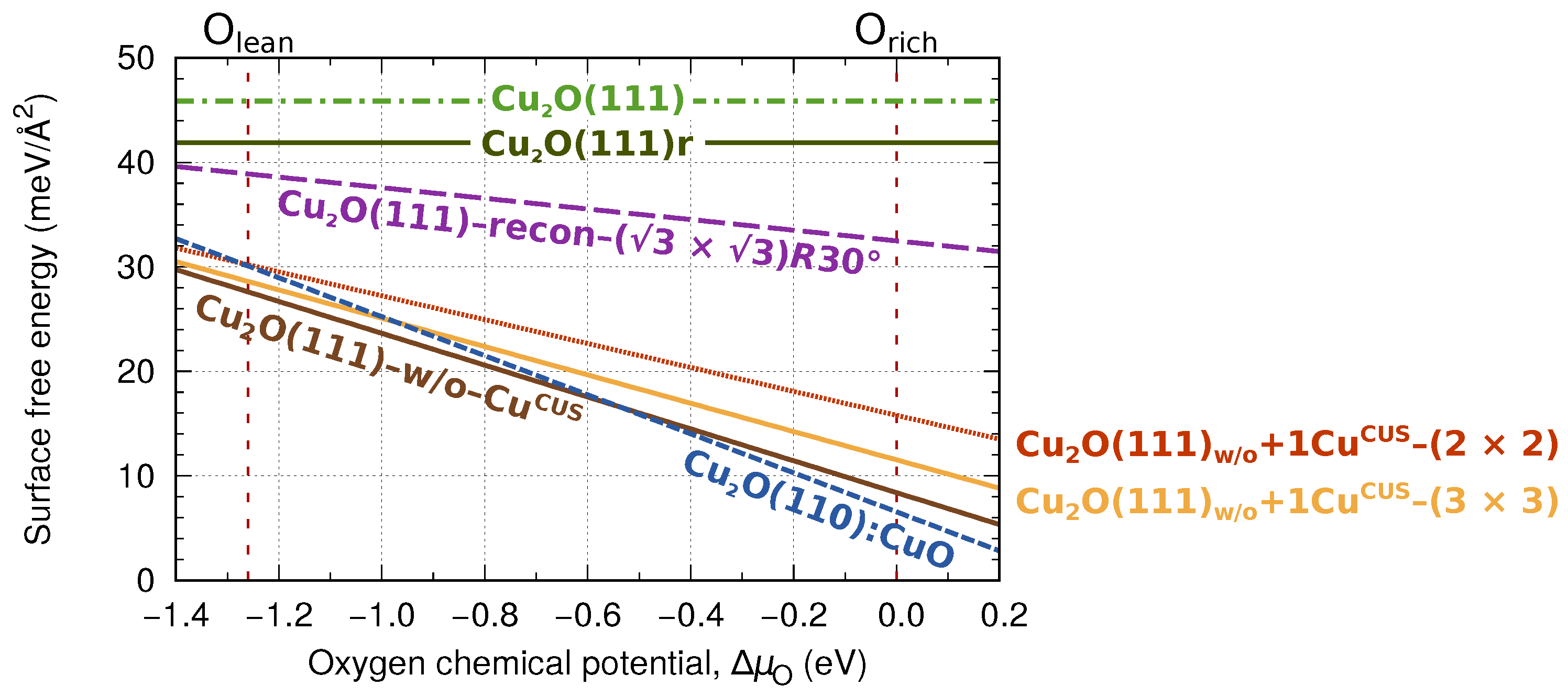
3.1. The Thermodynamic Stability of Bare CuO Surfaces
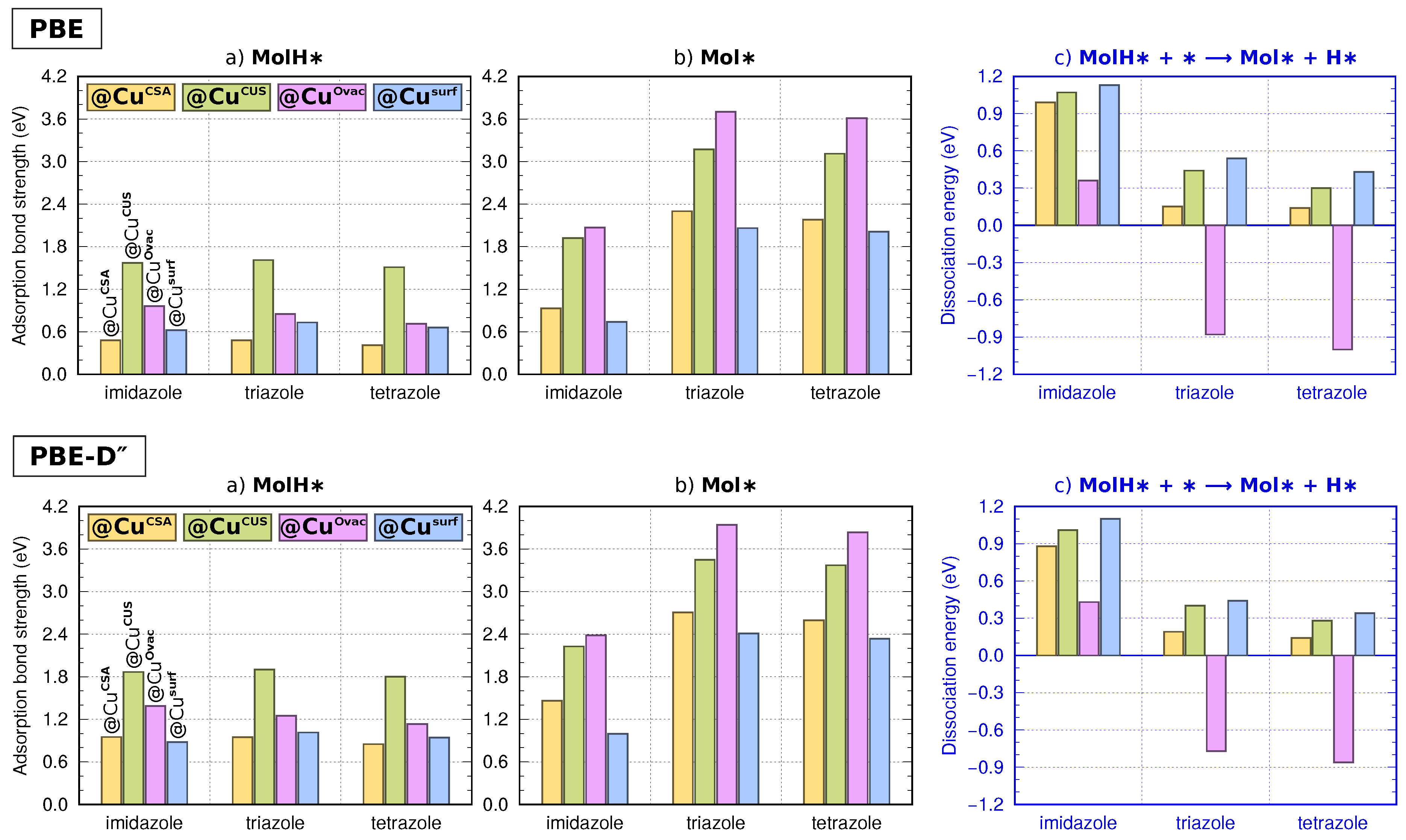
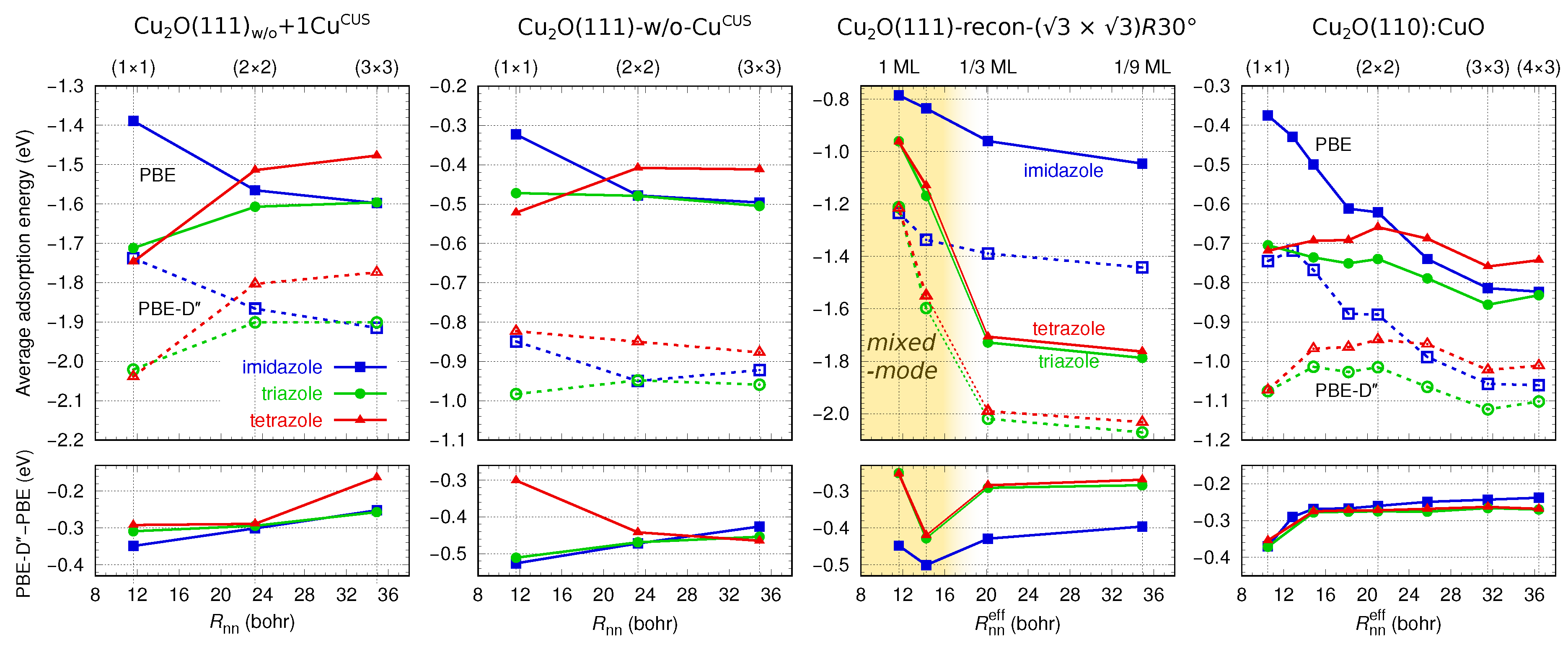
3.2. Molecular Adsorption at Lower Coverage
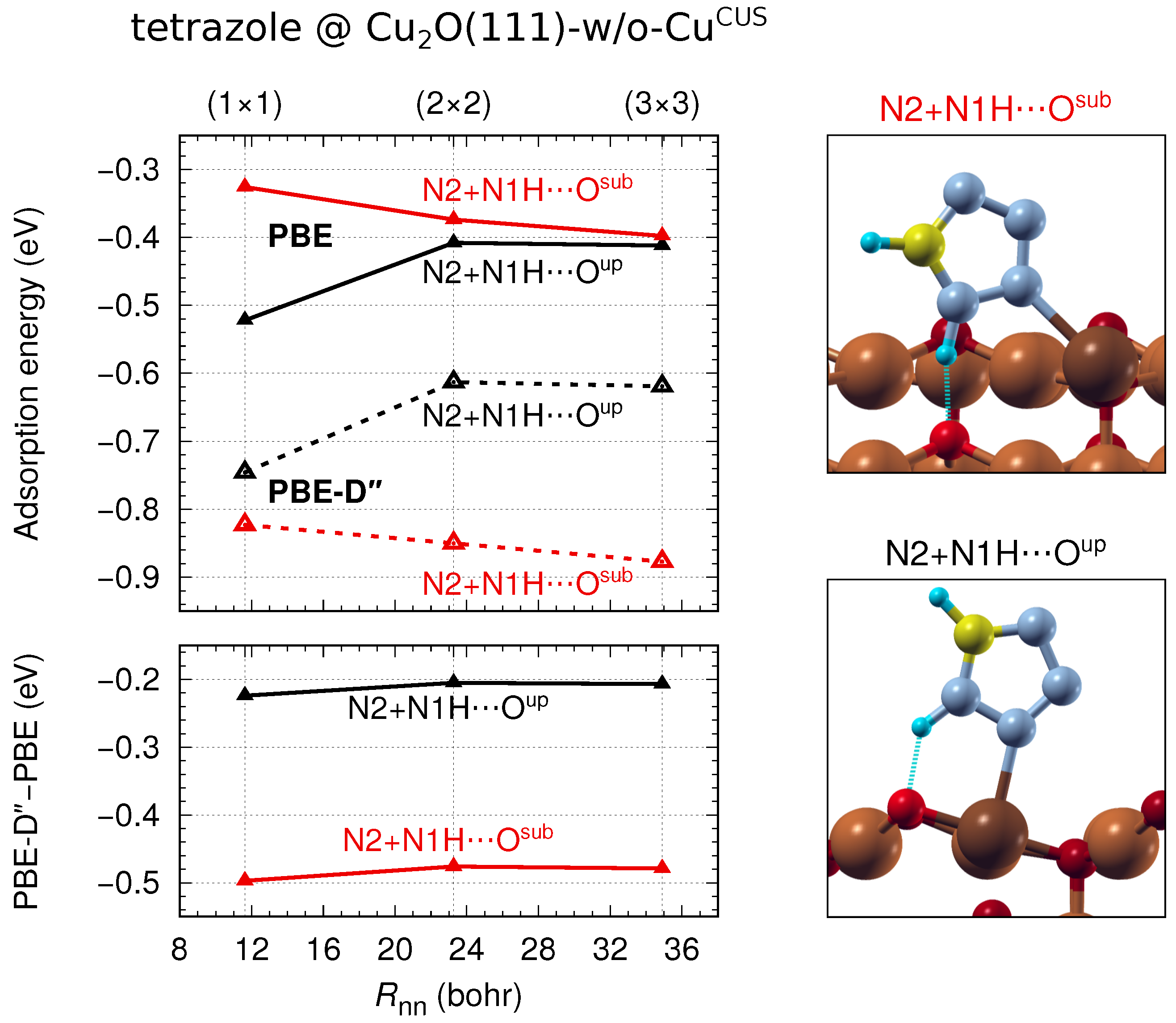
3.3. Lateral and Dispersion Interactions: PBE vs.
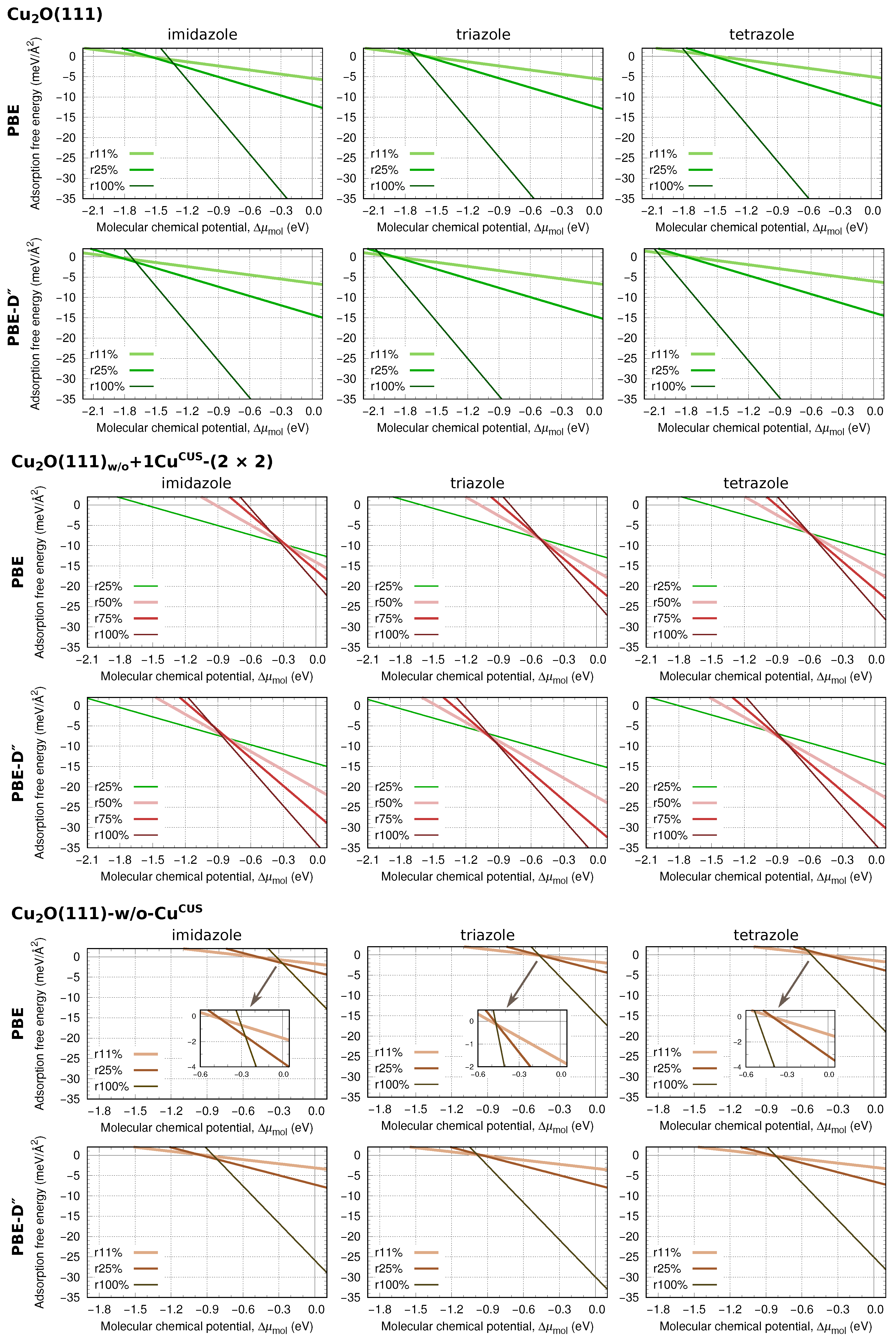
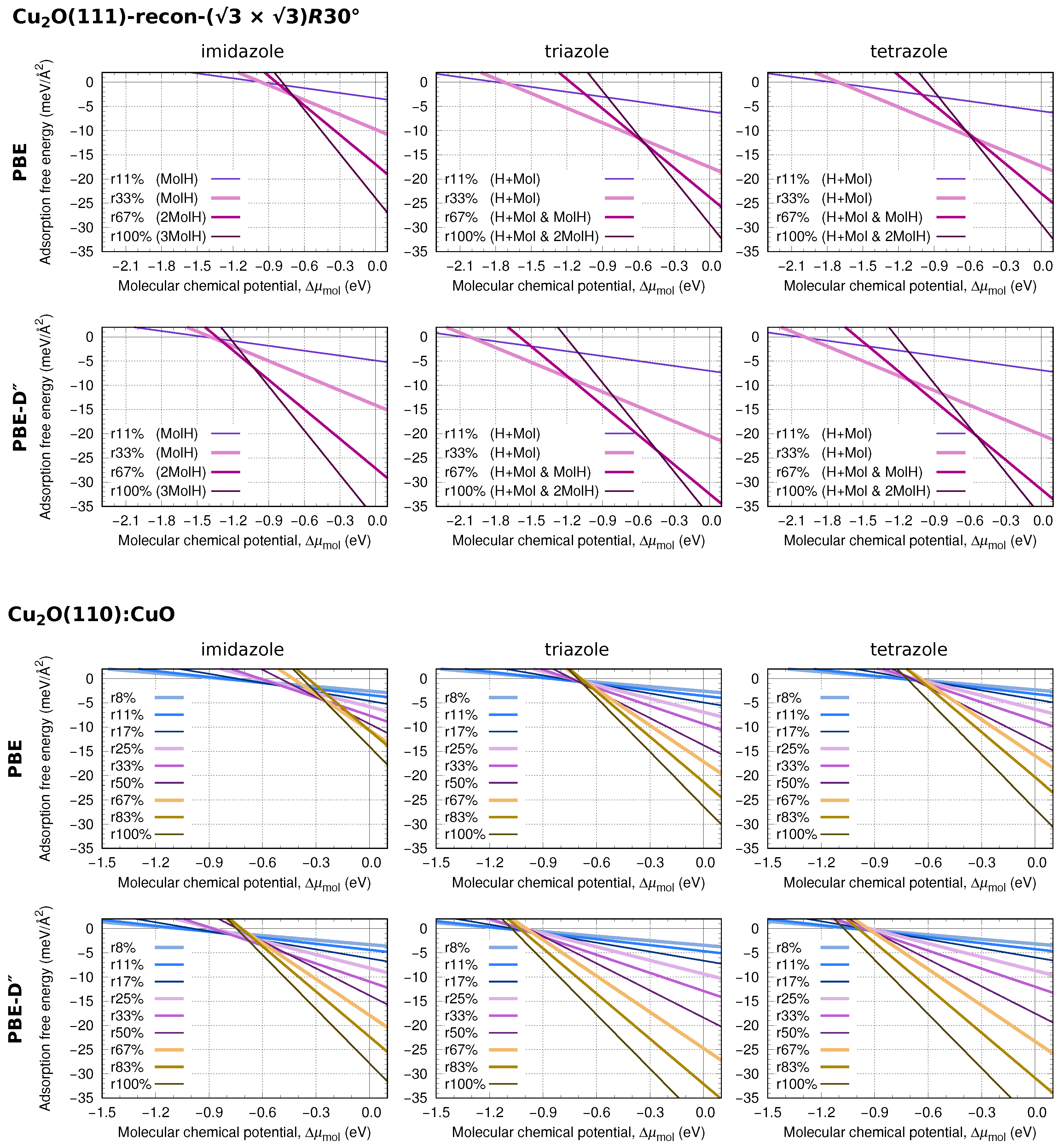
3.4. Thermodynamic Stability
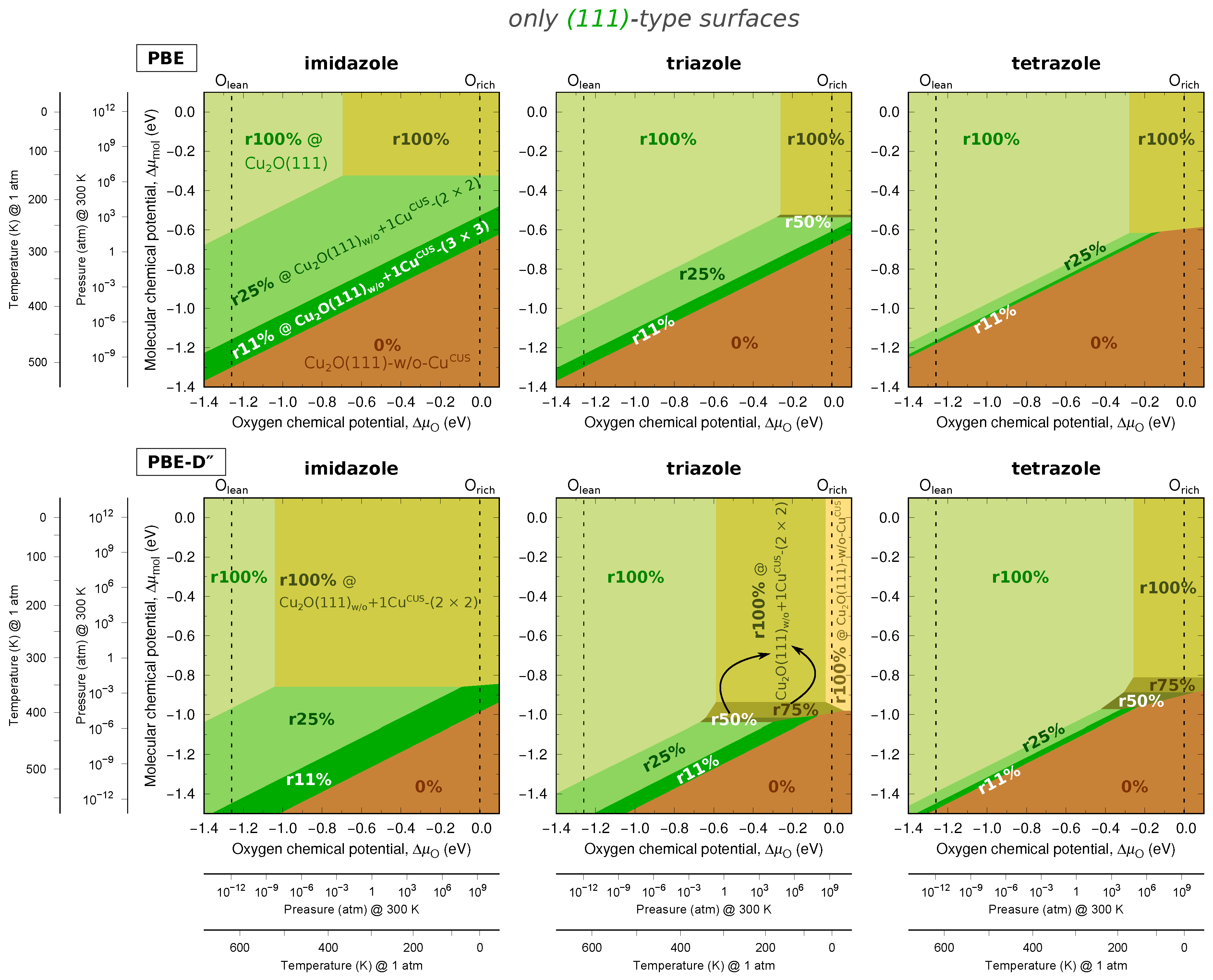
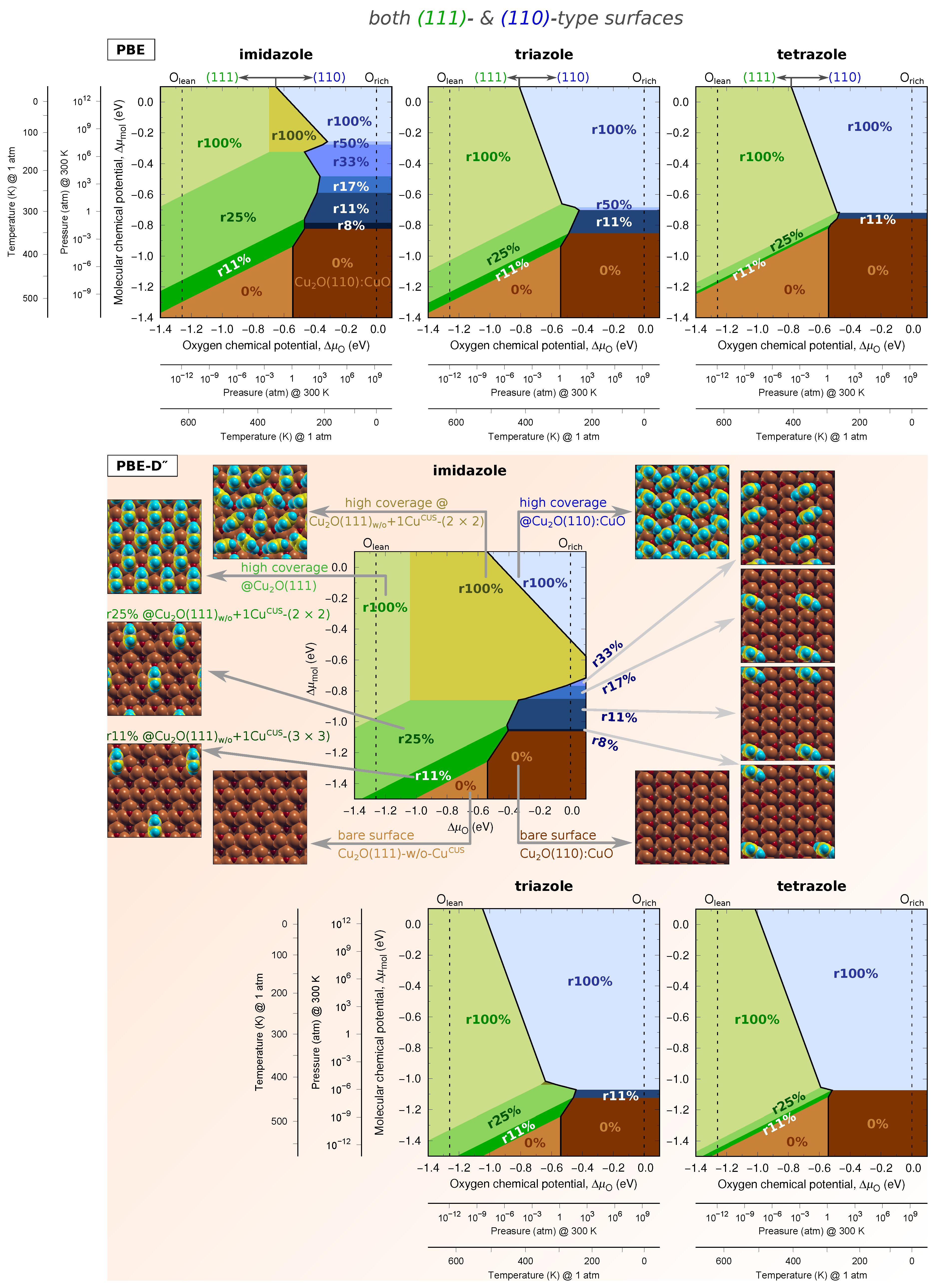
Two-Dimensional Phase Diagrams
4. Discussion
4.1. On the Role of CuCUS and CuOvac Sites
4.2. The Relevance of Current Results in the Context of Corrosion Inhibition
4.3. Drawbacks of the Approximate Ab Initio Thermodynamics Approach
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| GGA | generalized gradient approximation |
| PBE | Perdew–Burke–Ernzerhof |
| PBE with reparametrized empirical dispersion correction of Grimme | |
| BZ | Brillouin zone |
| CSA | coordinatively saturated |
| CUS | coordinatively unsaturated |
| abTD | ab initio thermodynamics |
Appendix A. Mapping of Molecular Chemical Potential into Temperature and Partial Pressure
Appendix B. Additional Figures





References
- Gustinčič, D.; Kokalj, A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding. Metals 2018, 8, 310. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure. Phys. Rev. B 2001, 65, 035406. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Isseroff, L.Y.; Carter, E.A. Importance of reference Hamiltonians containing exact exchange for accurate one-shot GW calculations of Cu2O. Phys. Rev. B 2012, 85, 235142. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Morgan, B.J.; Watson, G.W. Modeling the polaronic nature of p-type defects in Cu2O: The failure of GGA and GGA+U. J. Chem. Phys. 2009, 131, 124703. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Casarin, M.; Forrer, D.; Pavone, M.; Sambi, M.; Vittadini, A. Role and effective treatment of dispersive forces in materials: Polyethylene and graphite crystals as test cases. J. Comput. Chem. 2009, 30, 934–939. [Google Scholar] [CrossRef] [PubMed]
- McNellis, E.R.; Meyer, J.; Reuter, K. Azobenzene at coinage metal surfaces: Role of dispersive van der Waals interactions. Phys. Rev. B 2009, 80, 205414. [Google Scholar] [CrossRef]
- Kokalj, A.; Peljhan, S. Density functional theory study of ATA, BTAH, and BTAOH as copper corrosion inhibitors: Adsorption onto Cu(111) from gas phase. Langmuir 2010, 26, 14582–14593. [Google Scholar] [CrossRef] [PubMed]
- Tonigold, K.; Groß, A. Adsorption of small aromatic molecules on the (111) surfaces of noble metals: A density functional theory study with semiempirical corrections for dispersion effects. J. Chem. Phys. 2010, 132, 224701. [Google Scholar] [CrossRef] [PubMed]
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef] [PubMed]
- Gustinčič, D.; Kokalj, A. A DFT study of adsorption of imidazole, triazole, and tetrazole on oxidized copper surfaces: Cu2O(111) and Cu2O(111)-w/o-CuCUS. Phys. Chem. Chem. Phys. 2015, 17, 28602–28615. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Ultrasoft pseudopotentials for H, C, N, O, and Cu were taken from the Quantum Espresso Pseudopotential. 2017. Available online: http://www.quantum-espresso.org/pseudopotentials (accessed on 21 August 2017). (files: H.pbe-rrkjus.UPF, C.pbe-rrkjus.UPF, N.pbe-rrkjus.UPF, O.pbe-rrkjus.UPF and Cu.pbe-d-rrkjus.UPF).
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal contraction and disordering of the Al(110) surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A.; Peljhan, S. Density functional theory study of adsorption of benzotriazole on Cu2O surfaces. J. Phys. Chem. C 2015, 119, 11625–11635. [Google Scholar] [CrossRef]
- Bengtsson, L. Dipole correction for surface supercell calculations. Phys. Rev. B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Soon, A.; Todorova, M.; Delley, B.; Stampfl, C. Thermodynamic stability and structure of copper oxide surfaces: A first-principles investigation. Phys. Rev. B 2007, 75, 125420–125429. [Google Scholar] [CrossRef]
- Li, C.; Wang, F.; Li, S.; Sun, Q.; Jia, Y. Stability and electronic properties of the O-terminated Cu2O(111) surfaces: First-principles investigation. Phys. Lett. A 2010, 374, 2994–2998. [Google Scholar] [CrossRef]
- Önsten, A.; Göthelid, M.; Karlsson, U.O. Atomic structure of Cu2O(111). Surf. Sci. 2009, 603, 257–264. [Google Scholar] [CrossRef]
- Bendavid, L.I.; Carter, E.A. First-principles predictions of the structure, stability, and photocatalytic potential of Cu2O surfaces. J. Phys. Chem. B 2013, 117, 15750–15760. [Google Scholar] [CrossRef] [PubMed]
- Nilius, N.; Fedderwitz, H.; Groß, B.; Noguera, C.; Goniakowski, J. Incorrect DFT-GGA predictions of the stability of non-stoichiometric/polar dielectric surfaces: the case of Cu2O(111). Phys. Chem. Chem. Phys. 2016, 18, 6729–6733. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A. Ab initio modeling of the bonding of benzotriazole corrosion inhibitor to reduced and oxidized copper surfaces. Faraday Discuss. 2015, 180, 415–438. [Google Scholar] [CrossRef] [PubMed]
- Besharat, Z.; Stenlid, J.H.; Soldemo, M.; Marks, K.; Önsten, A.; Johnson, M.; Öström, H.; Weissenrieder, J.; Brinck, T.; Göthelid, M. Dehydrogenation of methanol on Cu2O(100) and (111). J. Chem. Phys. 2017, 146, 244702. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, N.; Kokalj, A. DFT Study of Interaction of Azoles with Cu(111) and Al(111) Surfaces: Role of Azole Nitrogen Atoms and Dipole–Dipole Interactions. J. Phys. Chem. C 2011, 115, 24189–24197. [Google Scholar] [CrossRef]
- Kokalj, A. Electrostatic model for treating long-range lateral interactions between polar molecules adsorbed on metal surfaces. Phys. Rev. B 2011, 84, 045418. [Google Scholar] [CrossRef]
- Peljhan, S.; Kokalj, A. DFT study of gas-phase adsorption of benzotriazole on Cu(111), Cu(100), Cu(110), and low coordinated defects thereon. Phys. Chem. Chem. Phys. 2011, 13, 20408–20417. [Google Scholar] [CrossRef] [PubMed]
- Peljhan, S.; Koller, J.; Kokalj, A. The Effect of Surface Geometry of Copper on Adsorption of Benzotriazole and Cl. Part I. J. Phys. Chem. C 2014, 118, 933–943. [Google Scholar] [CrossRef]
- Kovačević, N.; Milošev, I.; Kokalj, A. The roles of mercapto, benzene, and methyl groups in the corrosion inhibition of imidazoles on copper: II. Inhibitor–copper bonding. Corros. Sci. 2015, 98, 457–470. [Google Scholar] [CrossRef]
- Soldemo, M.; Stenlid, J.H.; Besharat, Z.; Johansson, N.; Önsten, A.; Knudsen, J.; Schnadt, J.; Göthelid, M.; Brinck, T.; Weissenrieder, J. Interaction of sulfur dioxide and near-ambient pressures of water vapor with cuprous oxide surfaces. J. Phys. Chem. C 2017, 121, 24011–24024. [Google Scholar] [CrossRef]
- Deng, X.; Herranz, T.; Weis, C.; Bluhm, H.; Salmeron, M. Adsorption of Water on Cu2O and Al2O3 Thin Films. J. Phys. Chem. C 2008, 112, 9668–9672. [Google Scholar] [CrossRef]
- Kokalj, A.; Costa, D. Molecular Modeling of Corrosion Inhibitors. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 6.1, pp. 332–345. [Google Scholar] [CrossRef]
- Kuznetsov, Y.; Kazansky, L. Physicochemical aspects of metal protection by azoles as corrosion inhibitors. Russ. Chem. Rev. 2008, 77, 219–232. [Google Scholar] [CrossRef]
- Milošev, I.; Kovačević, N.; Kovač, J.; Kokalj, A. The roles of mercapto, benzene and methyl groups in the corrosion inhibition of imidazoles on copper: I. Experimental characterization. Corros. Sci. 2015, 98, 107–118. [Google Scholar] [CrossRef]
- Kovačević, N.; Milošev, I.; Kokalj, A. How relevant is the adsorption bonding of imidazoles and triazoles for their corrosion inhibition of copper? Corros. Sci. 2017, 124, 25–34. [Google Scholar] [CrossRef]
- Schmickler, W.; Santos, E. Interfacial Electrochemistry, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Poberžnik, M.; Costa, D.; Hemeryck, A.; Kokalj, A. Insight into the Bonding of Silanols to Oxidized Aluminum Surfaces. J. Phys. Chem C 2018, in press. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustinčič, D.; Kokalj, A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability. Metals 2018, 8, 311. https://doi.org/10.3390/met8050311
Gustinčič D, Kokalj A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability. Metals. 2018; 8(5):311. https://doi.org/10.3390/met8050311
Chicago/Turabian StyleGustinčič, Dunja, and Anton Kokalj. 2018. "DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability" Metals 8, no. 5: 311. https://doi.org/10.3390/met8050311
APA StyleGustinčič, D., & Kokalj, A. (2018). DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability. Metals, 8(5), 311. https://doi.org/10.3390/met8050311