
Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity



Abstract
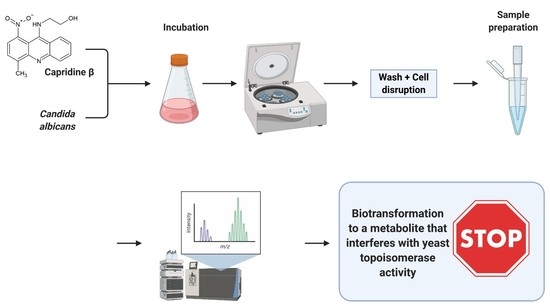
:
1. Introduction
2. Results and Discussion
2.1. Antifungal Activity
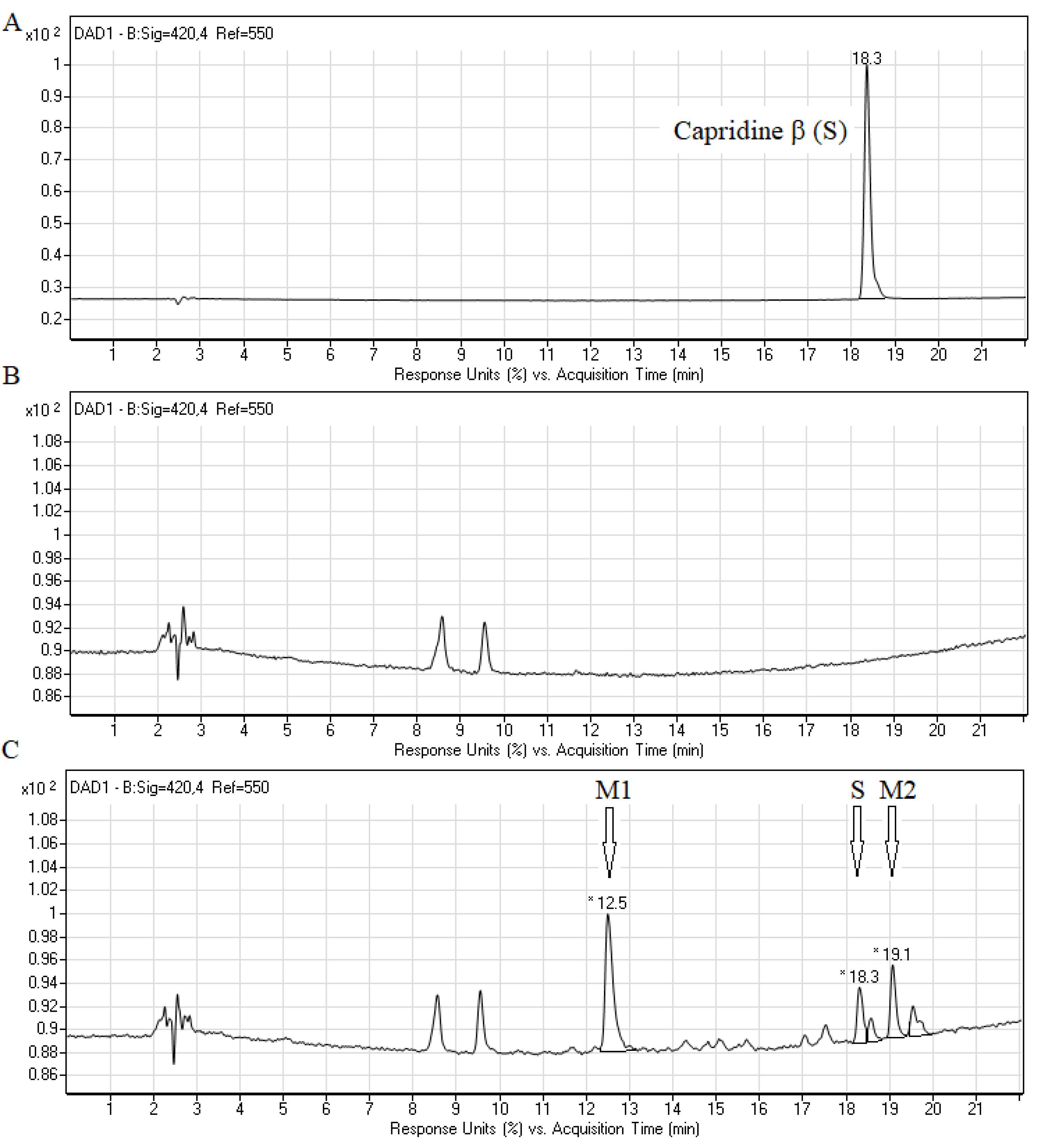
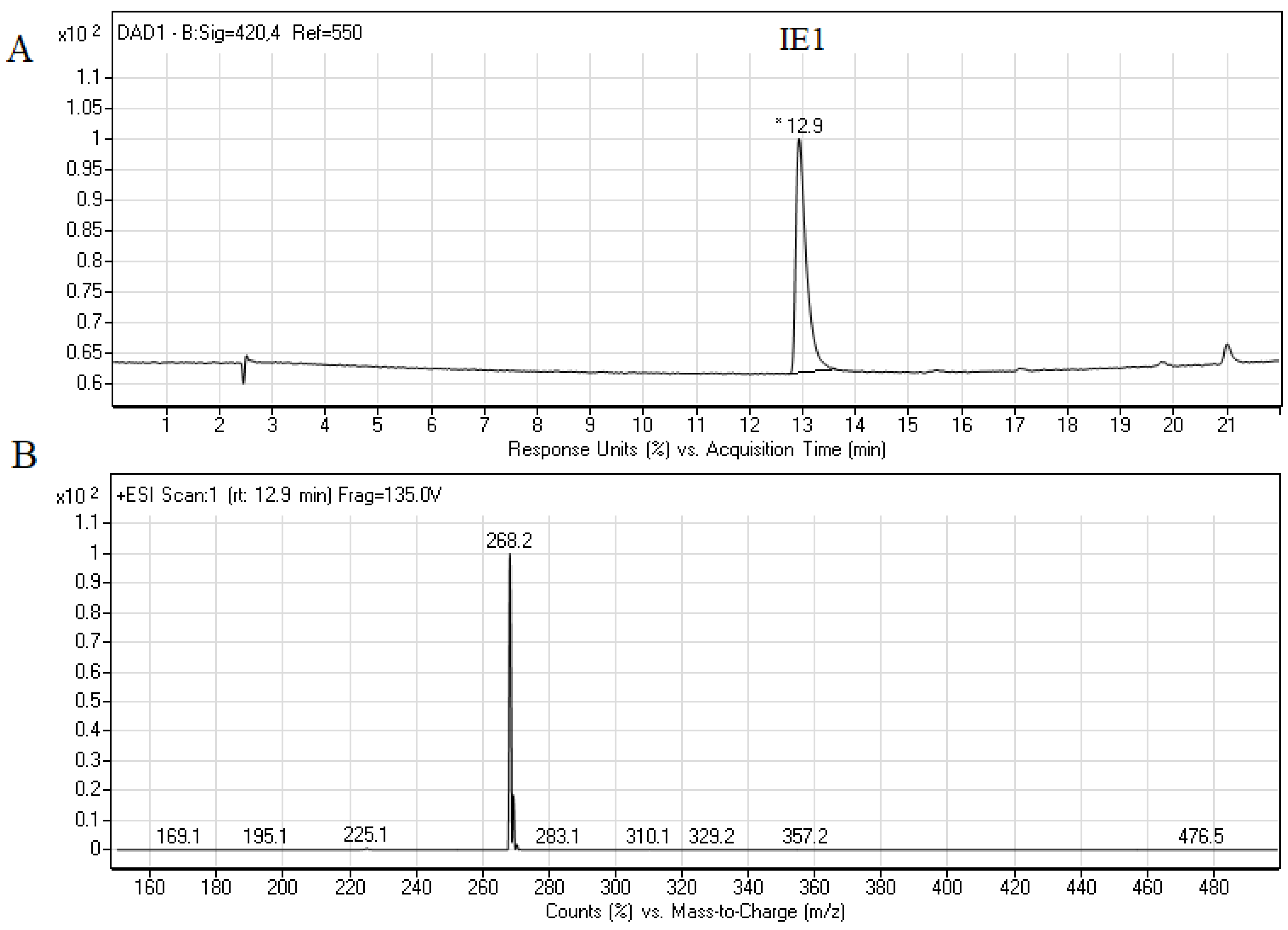
2.2. Biotransformation of Capridine β in Fungal Cells
2.3. Biological Activity of Capridine β Reduced Form (IE1), Identified as M1 Metabolite
3. Materials and Methods
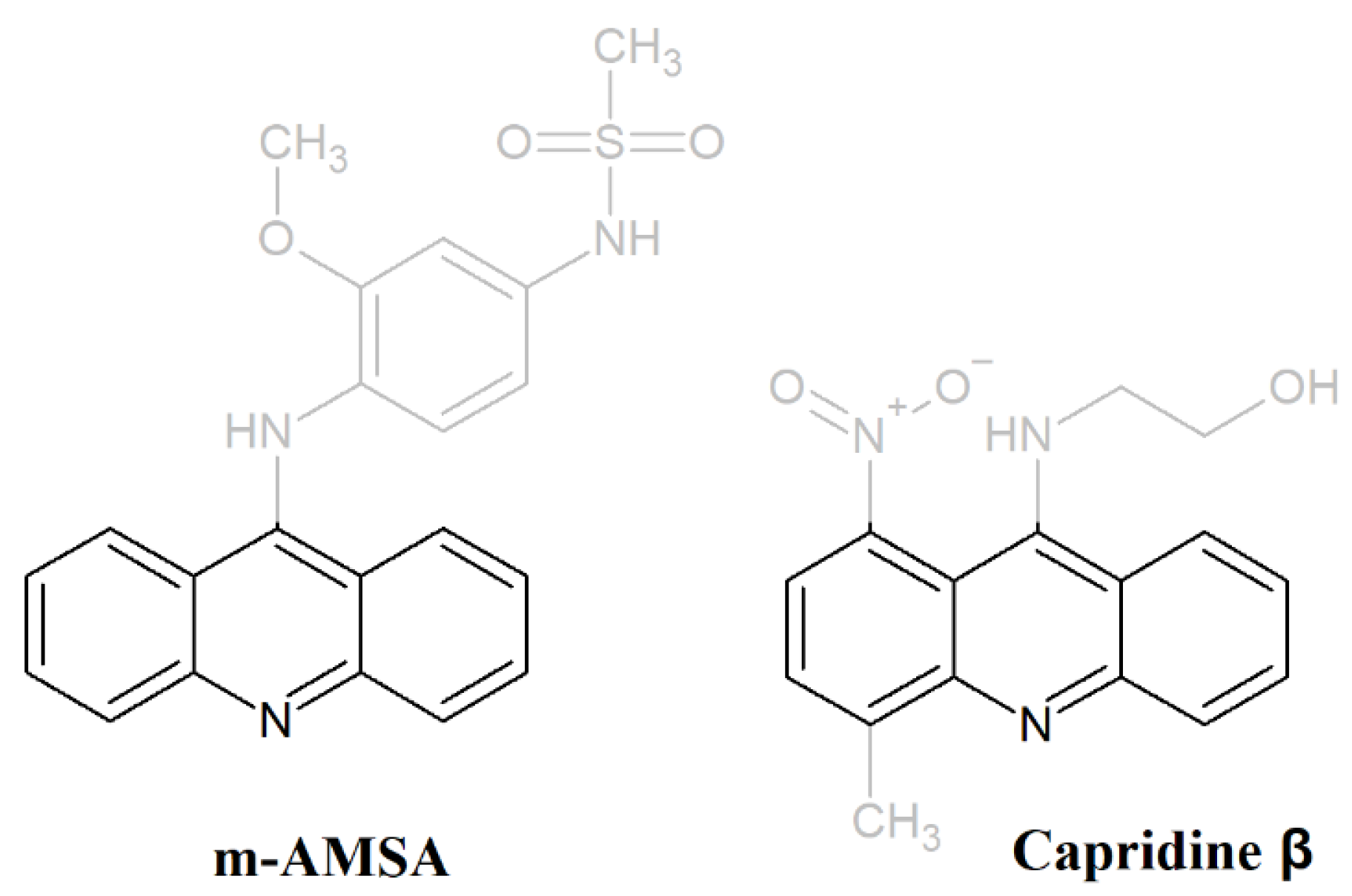
3.1. Chemical Synthesis of Capridine β and IE1
3.1.1. General
3.1.2. The Synthesis of Capridine β
3.1.3. The Synthesis of IE1
3.2. Microorganisms Strains and Growth Conditions
3.3. Metabolism Assays with C. albicans Cells
3.4. Chromatographic Analysis
3.5. Antimicrobial Activity Assay
3.6. Acridine Derivatives Accumulation in Microbial Cells
3.7. Yeast Topoisomerase II Relaxation Assay and Inhibition
3.8. UV-Vis and Fluorescence Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wainwright, M. Acridine-a neglected antibacterial chromophore. J. Antimicrob. Chemother. 2001, 47, 1–13. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, I. “Acridines” as new horizons in antifungal treatment. Molecules 2020, 25, 1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Li, T.-K.; Houghton, P.J.; Desai, S.D.; Daroui, P.; Liu, A.A.; Hars, E.S.; Ruchelman, A.L.; Lavoie, E.J.; Liu, L.F. Characterization of ARC-111 as a novel topoisomerase I-targeting anticancer drug. Cancer Res. 2003, 63, 8400–8407. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, S.; Voelkel, K.; Sternglanz, R. DNA topoisomerase II mutant of Saccharomyces cerevisiae: Topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc. Natl. Acad. Sci. USA 1984, 81, 2616–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, C.; Stearns, T.; Botstein, D. DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol. Cell. Biol. 1989, 9, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Kwok, S.C.; Schelenz, S.; Wang, X.; Steverding, D. In Vitro effect of DNA topoisomerase inhibitors on Candida albicans. Med. Mycol. 2010, 48, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.I.; Nimrod, A.C.; Mehrpooya, M.; Nitiss, J.L.; Walker, L.A.; Clark, A.M. Antifungal activity of eupolauridine and its action on DNA topoisomerases. Antimicrob. Agents Chemother. 2002, 46, 1845–1850. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.L.; Baranowski, J.; Fostel, J.; Montgomery, D.A.; Lartey, P.A. DNA topoisomerases from pathogenic fungi: Targets for the discovery of antifungal drugs. Antimicrob. Agents Chemother. 1992, 36, 2778–2784. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, M.; Toffaletti, D.L.; Rude, T.H.; Dykstra, C.C.; Heitman, J.; Perfect, J.R. Topoisomerase I is essential in Cryp-tococcus neoformans: Role in pathobiology and as an antifungal target. Genetics 1999, 152, 167–178. [Google Scholar] [PubMed]
- Jiang, W.; Gerhold, D.; Kmiec, E.B.; Hauser, M.; Becker, J.M.; Koltin, Y. The topoisomerase I gene from Candida albicans. Microbiology 1997, 143, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, E.; Moś-Rompa, A.; Nowak-Ziatyk, D.; Konopa, J. Antitumor 1-nitroacridine derivative C-1748, induces apoptosis, necrosis or senescence in human colon carcinoma HCT8 and HT29 cells. Biochem. Pharm. 2010, 79, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewska, A.; Niemira, M.; Jagiełło, K.; Potęga, A.; Świst, M.; Henderson, C.; Skwarska, A.; Augustin, E.; Konopa, J.; Mazerska, Z.; et al. Diminished toxicity of C-1748, 4-methyl-9-hydroxyethylamino-1-nitroacridine, compared with its demethyl analog, C-857, corresponds to its resistance to metabolism in HepG2 cells. Biochem. Pharm. 2012, 84, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Rząd, K.; Paluszkiewicz, E.; Gabriel, I. A new 1-nitro-9-aminoacridine derivative targeting yeast topoisomerase II able to overcome fluconazole-resistance. Bioorganic Med. Chem. Lett. 2021, 35, 127815. [Google Scholar] [CrossRef]
- Tadi, K.; Ashok, B.T.; Chen, Y.; Banerjee, D.; Wysocka-Skrzela, B.; Konopa, J.; Darzynkiewicz, Z.; Tiwari, R.K. Pre-clinical evaluation of 1-nitroacridine derived chemotherapeutic agent that has preferential cytotoxic activity towards prostate cancer. Cancer Biol. 2007, 6, 1632–1637. [Google Scholar] [CrossRef] [Green Version]
- Ashok, B.; Tadi, K.; Banerjee, D.; Konopa, J.; Iatropoulos, M.; Tiwari, R.K. Pre-clinical toxicology and pathology of 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C-1748), a novel anti-cancer agent in male Beagle dogs. Life Sci. 2006, 79, 1334–1342. [Google Scholar] [CrossRef]
- Ashok, B.T.; Tadi, K.; Garikapaty, V.P.; Chen, Y.; Huang, Q.; Banerjee, D.; Konopa, J.; Tiwari, R.K. Preclinical toxicological examination of a putative prostate cancer-specific 4-methyl-1-nitroacridine derivative in rodents. Anti-Cancer Drugs 2007, 18, 87–94. [Google Scholar] [CrossRef]
- Augustin, E.; Niemira, M.; Hołownia, A.; Mazerska, Z. CYP3A4-dependent cellular response does not relate to CYP3A4-catalysed metabolites of C-1748 and C-1305 acridine antitumor agents in HepG2 cells. Cell Biol. Int. 2014, 38, 1291–1303. [Google Scholar] [CrossRef]
- Tan, X.; Fuchs, B.B.; Wang, Y.; Chen, W.; Yuen, G.J.; Chen, R.B.; Jayamani, E.; Anastassopoulou, C.; Pukkila-Worley, R.; Coleman, J.J.; et al. The Role of Candida albicans SPT20 in filamentation, biofilm formation and pathogenesis. PLoS ONE 2014, 9, e94468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszek, A.; Konopa, J. 32P-post-labeling analysis of DNA adduct formation by antitumor drug nitracrine (Ledakrin) and other nitroacridines in different biological systems. Biochem. Pharm. 1989, 38, 1301–1312. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Taraszkiewicz, A.; Grinholc, M.; Bielawski, K.P.; Kawiak, A.; Nakonieczna, J. Imidazoacridinone derivatives as efficient sensitizers in photoantimicrobial chemotherapy. Appl. Environ. Microbiol. 2013, 79, 3692–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Li, Y.-C.; Wang, Y.-R.; Li, T.-K.; Chan, N.-L. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 2013, 41, 10630–10640. [Google Scholar] [CrossRef]
- Lemke, K.; Wojciechowski, M.; Laine, W.; Bailly, C.; Colson, P.; Baginski, M.; Larsen, A.K.; Skladanowski, A. Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res. 2005, 33, 6034–6047. [Google Scholar] [CrossRef] [Green Version]
- Mazerska, Z.; Sowiński, P.; Konopa, J. Molecular mechanism of the enzymatic oxidation investigated for imidazoacridinone antitumor drug, C-1311. Biochem. Pharm. 2003, 66, 1727–1736. [Google Scholar] [CrossRef]
- Konopa, J.; Wysocka-Skrzela, B.; Tiwari, R.K. 9-Alkilamino-1-Nitroacridine Derivatives; U.S. Patent 6.589.961 B2; Patent and Trademark Office: Alexandria, VA, USA, 2003.
- Wysocka-Skrzela, B. Research on tumor-inhibiting compounds: Reactions of 1-nitro-9-aminoacridine derivatives, new antitumor agents, with nucleophiles. Pol. J. Chem. 1986, 60, 317–318. [Google Scholar]
- CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Clinical and Laboratory Standard Institute: Annapolis Junction, MD, USA, 2008. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | * MIC90 μg mL−1 | ||||
|---|---|---|---|---|---|
| Candida albicans ATCC 10231 | Candida glabrata ATCC 90030 | Candida krusei ATCC 6258 | Candida parapsilosis ATCC 22019 | Saccharomyces cerevisiae ATCC 9763 | |
| Capridine β | 1 | 8 | 8 | 64 | 0.5 |
| IE1 | >64 | 64 | 64 | 64 | 64 |
| m-AMSA | >64 | >64 | >64 | >64 | >64 |
| Amphotericin B | 0.5 | 1 | 1 | 1 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabriel, I.; Rząd, K.; Paluszkiewicz, E.; Kozłowska-Tylingo, K. Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity. Pathogens 2021, 10, 189. https://doi.org/10.3390/pathogens10020189
Gabriel I, Rząd K, Paluszkiewicz E, Kozłowska-Tylingo K. Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity. Pathogens. 2021; 10(2):189. https://doi.org/10.3390/pathogens10020189
Chicago/Turabian StyleGabriel, Iwona, Kamila Rząd, Ewa Paluszkiewicz, and Katarzyna Kozłowska-Tylingo. 2021. "Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity" Pathogens 10, no. 2: 189. https://doi.org/10.3390/pathogens10020189
APA StyleGabriel, I., Rząd, K., Paluszkiewicz, E., & Kozłowska-Tylingo, K. (2021). Antifungal Activity of Capridine β as a Consequence of Its Biotransformation into Metabolite Affecting Yeast Topoisomerase II Activity. Pathogens, 10(2), 189. https://doi.org/10.3390/pathogens10020189