
The Emerging Concern and Interest SARS-CoV-2 Variants

 ,
,
Abstract
:1. Introduction
2. Variant of Concern
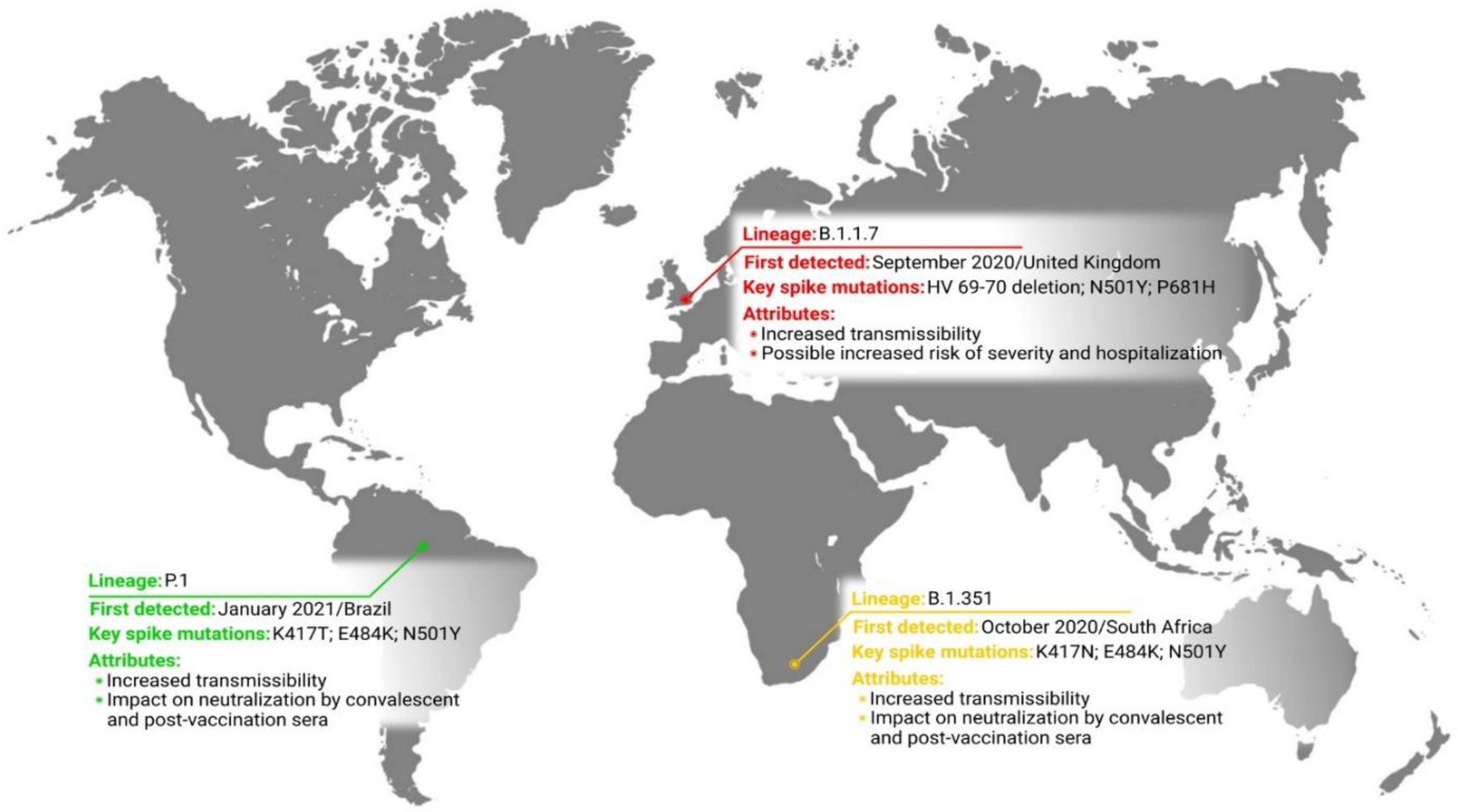
2.1. Lineage B.1.1.7
2.2. Lineage B.1.351
2.3. Lineage P.1
3. Variant of Interest
4. Variant of High Consequence
5. Mutations Impact on SARS-CoV-2 Diagnostics/Detection Protocols
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 3 March 2021).
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529. [Google Scholar] [CrossRef]
- Almubaid, Z.; Al-Mubaid, H. Analysis and comparison of genetic variants and mutations of the novel coronavirus SARS-CoV-2. Gene Rep. 2021, 23, 101064. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Petrone, M.E.; Holmes, E.C. We shouldn’t worry when a virus mutates during disease outbreaks. Nat. Microbiol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Ferron, F.; Subissi, L.; De Morais, A.T.S.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Walensky, R.P.; Walke, H.T.; Fauci, A.S. SARS-CoV-2 Variants of Concern in the United States—Challenges and Opportunities. JAMA 2021, 325, 1037. [Google Scholar] [CrossRef]
- Carrasco-Hernandez, R.; Jácome, R.; Vidal, Y.L.; De León, S.P. Are RNA Viruses Candidate Agents for the Next Global Pandemic? A Review. ILAR J. 2017, 58, 343–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 534, 374–380. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- World Health Organization. An Update on SARS-CoV-2 Virus Mutations & Variants. Available online: https://www.who.int/docs/default-source/coronaviruse/risk-comms-updates/update47-sars-cov-2-variants.pdf?sfvrsn=f2180835_4 (accessed on 25 March 2021).
- Centers for Disease Control and Prevention. About Variants of the Virus that Causes COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant.html (accessed on 25 March 2021).
- Burki, T. Understanding variants of SARS-CoV-2. Lancet 2021, 397, 462. [Google Scholar] [CrossRef]
- Centers for Disease and Control Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html#Interest (accessed on 24 March 2021).
- Lineages, P. Global Report Investigating Novel Coronavirus Haplotypes. 2021 Copyright© 2021 Massachusetts Medical Society. Available online: https://cov-lineages.org/global_report (accessed on 24 March 2021).
- World Health Organization. Weekly Epidemiological Update on COVID-19—23 March 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---23-march-2021 (accessed on 24 March 2021).
- Huang, S.-W.; Miller, S.O.; Yen, C.-H.; Wang, S.-F. Impact of Genetic Variability in ACE2 Expression on the Evolutionary Dynamics of SARS-CoV-2 Spike D614G Mutation. Genes 2020, 12, 16. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, 74, 13525. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. SARS-CoV-2 Genomic Sequencing for Public Health Goals. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-genomic_sequencing-2021.1 (accessed on 10 May 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Weekly Epidemiological Update. Special Edition: Proposed Working Definitions of SARS-CoV-2 Variants of Interest and Variants of Concern. Available online: https://www.who.int/Publications/m/item/covid-19-weekly-epidemiological-update (accessed on 24 March 2021).
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020–January 12, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: Matched cohort study. BMJ 2021, 372. [Google Scholar] [CrossRef]
- Grint, D.J.; Wing, K.; Williamson, E.; McDonald, H.I.; Bhaskaran, K.; Evans, D.; Evans, S.J.; Walker, A.J.; Hickman, G.; Nightingale, E.; et al. Case fatality risk of the SARS-CoV-2 variant of concern B.1.1.7 in England, 16 November to 5 February. Eurosurveillance 2021, 26, 2100256. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. US COVID-19 Cases Caused by Variants. Available online: https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant-cases.html (accessed on 22 March 2021).
- Global Report Investigating Novel Coronavirus Haplotypes. B.1.1.7. Available online: https://cov-lineages.org/global_report_B.1.1.7.html#table2link (accessed on 23 March 2021).
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 18 March 2021).
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nat. Cell Biol. 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Gu, H.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.-Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef]
- Santos, J.C.; Passos, G.A. The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pereira, F. SARS-CoV-2 variants combining spike mutations and the absence of ORF8 may be more transmissible and require close monitoring. Biochem. Biophys. Res. Commun. 2021, 550, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe 2021, 29, 529–539.e3. [Google Scholar] [CrossRef]
- Graham, M.S.; Sudre, C.H.; May, A.; Antonelli, M.; Murray, B.; Varsavsky, T.; Kläser, K.; Canas, L.S.; Molteni, E.; Modat, M.; et al. Changes in symptomatology, reinfection, and transmissibility associated with the SARS-CoV-2 variant B.1.1.7: An ecological study. Lancet Public Health 2021, 6, e335–e345. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nat. Cell Biol. 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Calistri, P.; Amato, L.; Puglia, I.; Cito, F.; Di Giuseppe, A.; Danzetta, M.L.; Morelli, D.; Di Domenico, M.; Caporale, M.; Scialabba, S.; et al. Infection sustained by lineage B.1.1.7 of SARS-CoV-2 is characterised by longer persistence and higher viral RNA loads in nasopharyngeal swabs. Int. J. Infect. Dis. 2021, 105, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.P.; Walls, A.C.; Bassi, J.; Pinto, D.; Fregni, C.S. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nat. Cell Biol. 2021, 593, 136–141. [Google Scholar] [CrossRef]
- Wu, K.; Werner, A.P.; Moliva, J.I.; Koch, M.; Choi, A.; Stewart-Jones, G.B.E.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; Graham, B.S. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. BioRxiv 2021. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.-K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; Türeci, Ö.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine–elicited human sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef]
- Mwenda, M.; Saasa, N.; Sinyange, N.; Busby, G.; Chipimo, P.J.; Hendry, J.; Kapona, O.; Yingst, S.; Hines, J.Z.; Minchella, P.; et al. Detection of B.1.351 SARS-CoV-2 Variant Strain—Zambia, December 2020. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 280–282. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Science Brief: Emerging SARS-CoV-2 Variants. Available online: https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html (accessed on 22 March 2021).
- European Centre for Disease Prevention and Control. SARS-CoV-2 Increased Circulation of Variants of Concern and Vaccine Rollout in the EU/EEA, 14th Update. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/RRA-covid-19-14th-update-15-feb-2021.pdf (accessed on 22 March 2021).
- Global Report Investigating Novel Coronavirus Haplotypes. B.1.35. Available online: https://cov-lineages.org/global_report_B.1.351.html (accessed on 23 March 2021).
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cele, S.; Gazy, I.; Jackson, L.; Hwa, S.-H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; Naidoo, Y.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nat. Cell Biol. 2021, 593, 142–146. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nat. Cell Biol. 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Lopez-Rincon, A.; Perez-Romero, C.A.; Tonda, A.; Mendoza-Maldonado, L.; Claassen, E.; Garssen, J.; Kraneveld, A.D. Design of Specific Primer Sets for the Detection of B.1.1.7, B.1.351 and P.1 SARS-CoV-2 Variants using Deep Learning. bioRxiv 2021. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Huang, S.-W.; Wang, S.-F. SARS-CoV-2 Entry Related Viral and Host Genetic Variations: Implications on COVID-19 Severity, Immune Escape, and Infectivity. Int. J. Mol. Sci. 2021, 22, 3060. [Google Scholar] [CrossRef]
- Faria, N.R.; Claro, I.M.; Candido, D.; Moyses Franco, L.A.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: Preliminary findings. J. Virol. 2021, 12. [Google Scholar] [CrossRef]
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant Identified in Travelers from Brazil to Japan. Emerg. Infect. Dis. 2021, 27, 1243–1245. [Google Scholar] [CrossRef]
- Firestone, M.J.; Lorentz, A.J.; Meyer, S.; Wang, X.; Como-Sabetti, K.; Vetter, S.; Smith, K.; Holzbauer, S.; Beaudoin, A.; Garfin, J.; et al. First Identified Cases of SARS-CoV-2 Variant P.1 in the United States—Minnesota, January 2021. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Global Report Investigating Novel Coronavirus Haplotypes. P.1. Available online: https://cov-lineages.org/global_report_P.1.html (accessed on 23 March 2021).
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, eabh2644. [Google Scholar] [CrossRef]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A., Jr.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; da Silva Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased Resistance of SARS-CoV-2 Variant P.1 to Antibody Neutralization. bioRxiv 2021. [Google Scholar] [CrossRef]
- De Souza, W.M.; Amorim, M.R.; Sesti-Costa, R.; Coimbra, L.D.; de Toledo-Teixeira, D.A.; Parise, P.L.; Barbosa, P.P.; Bispo-Dos-Santos, K.; Mofatto, L.S.; Simeoni, C.L.; et al. Levels of SARS-CoV-2 Lineage P.1 Neutralization by Antibodies Elicited after Natural Infection and Vaccination. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Naveca, F.; da Costa, C.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Costa, Á.; Duarte, D.; Silva, G.; Mejía, M. SARS-CoV-2 Reinfection by the New Variant of Concern (VOC) P.1 in Amazonas, Brazil. Virological. Org. Preprint. Available online: https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/5962021 (accessed on 24 March 2021).
- PANGO Lineages. Lineage B.1.526. Available online: https://cov-lineages.org/lineages/lineage_B.1.526.html (accessed on 24 March 2021).
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Böger, B.; Fachi, M.M.; Vilhena, R.O.; Cobre, A.F.; Tonin, F.S.; Pontarolo, R. Systematic review with meta-analysis of the accuracy of diagnostic tests for COVID-19. Am. J. Infect. Control. 2021, 49, 21–29. [Google Scholar] [CrossRef]
- Wollschlaeger, P.; Gerlitz, N.; Todt, D.; Pfaender, S.; Bollinger, T.; Sing, A.; Dangel, A.; Ackermann, N.; Korn, K.; Ensser, A.; et al. SARS-CoV-2 N gene dropout and N gene Ct value shift as indicator for the presence of B.1.1.7 lineage in a widely used commercial multiplex PCR assay. medRxiv 2021. [Google Scholar] [CrossRef]
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.-P.; Bours, V.; et al. A Recurrent Mutation at Position 26340 of SARS-CoV-2 Is Associated with Failure of the E Gene Quantitative Reverse Transcription-PCR Utilized in a Commercial Dual-Target Diagnostic Assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Sundararaju, S.; Manickam, C.; Mirza, F.; Al-Hail, H.; Lorenz, S.; Tang, P. A Novel Point Mutation in the N Gene of SARS-CoV-2 May Affect the Detection of the Virus by Reverse Transcription-Quantitative PCR. J. Clin. Microbiol. 2021, 59, e03278-20. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Eurosurveillance 2020, 25, 2001650. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.; Brancaccio, G.; Brazzale, A.R.; Lavezzo, E.; Onelia, F.; Franchin, E.; Manuto, L.; Bianca, F.; Cianci, V.; Cattelan, A.; et al. Emergence of N antigen SARS-CoV-2 genetic variants escaping detection of antigenic tests. medRxiv 2021. [Google Scholar] [CrossRef]
- The U.S. Food and Drug Administration. SARS-CoV-2 Viral Mutations: Impact on COVID-19 Tests. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests (accessed on 12 May 2021).
- Zuckerman, N.S.; Fleishon, S.; Bucris, E.; Bar-Ilan, D.; Linial, M.; Bar-Or, I.; Indenbaum, V.; Weil, M.; Mendelson, E.; Mandelboim, M.; et al. A unique SARS-CoV-2 spike protein P681H strain detected in Israel. medRxiv 2021. [Google Scholar] [CrossRef]
- Statista. Rate of COVID-19 Vaccination Worldwide as of 31 March 2021, by Country or Territory*. Available online: https://www.statista.com/statistics/1194939/rate-covid-vaccination-by-county-worldwide/ (accessed on 1 April 2021).


{kind=link}
| Gene | Nucleotide | Amino Acid |
|---|---|---|
| ORF1ab | C3267T | T1001I |
| C5388A | A1708D | |
| T6954C | I2230T | |
| 11288–11296 deletion | SGF 3675–3677 deletion | |
| Spike | 21765–21770 deletion | HV 69–70 deletion (mutation of concern) |
| 21991–21993 deletion | Y144 deletion | |
| A23063T | N501Y (mutation of concern) | |
| C23271A | A570D | |
| C23604A | P681H (mutation of concern) | |
| C23709T | T716I | |
| T24506G | S982A | |
| G24914C | D1118H | |
| ORF8 | C27972T | Q27stop |
| G28048T | R52I | |
| A28111G | Y73C | |
| N | 28280 GAT- > CTA | D3L |
| C28977T | S235F |
| Gene | Nucleotide | Amino Acid |
|---|---|---|
| ORF1ab | C1059T | T265I |
| G5230T | K1655N | |
| C8660T | H2799Y | |
| C8964T | S2900L | |
| A10323G | K3353R | |
| G13843T | D4527Y | |
| C17999T | T5912I | |
| Spike | C21614T | L18F |
| A21801C | D80A | |
| A2206G | D215G | |
| 22286-22294 deletion | L242_244L deletion | |
| G22299T | R246I | |
| G22813T | K417N | |
| G23012A | E484K | |
| A23063T | N501Y | |
| C23664T | A701V | |
| ORF3a | G25563T | Q57H |
| C25904T | S71L | |
| E | C26456T | P71L |
| N | C28887T | T205I |
| Characteristic | B.1.1.7 | B.1.351 | P.1 |
|---|---|---|---|
| Characteristic spike mutations | HV 69/70 deletion, Y144 deletion, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H | L18F, D80A, D215G, L242_244L deletion, R246I, K417N, E484K, N501Y, D614G, A701V | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G H655Y, T1027I, V1176F |
| Transmissibility | Increased transmissibility (43% to 90% more transmissible than previously circulating variants). | Suggested increased transmissibility (50% more transmissible than the previously circulating variants). | Increased, 1.7–2.4-fold more transmissible than previous circulating variants |
| Severity | Possible increased risk of severity and mortality of illness. | No significant evidence of an impact on COVID-19 course severity or mortality. | No significant evidence of an impact on COVID-19 course severity or mortality. |
| Immune susceptibility | Refractory or moderately resistant to neutralization by monoclonal antibodies (mAbs). Not resistant to neutralization by convalescent plasma and vaccine sera. | Potential increased risk of reinfection. Resistant to neutralization by mAbs. Markedly resistant to neutralization by convalescent plasma and vaccine sera. | Potential increased risk of reinfection. Resistant to neutralization by mAbs. Notable loss of neutralizing activity by convalescent plasma and vaccine sera. |
| References | [32,33,34,39,42,43,44] | [45,46,47,51] | [47,60,62,63,64] |
| Name (PANGO Lineage) | Next Strain Clade | First Detected | Key Spike Mutations | Characteristic |
|---|---|---|---|---|
| B.1.526 | 20C | United States (November 2020) | T95I, D253G, L5F, S477N, E484K, D614G, A701V | Potential depletion in neutralization by convalescent and post-vaccination sera or monoclonal antibody treatments |
| B.1.525 | 20C | United States (December 2020) | H69-V70 deletion, Y144 deletion, Q52R; E484K, Q677H; D614G, F888L | Potential depletion in neutralization by convalescent and post-vaccination sera or monoclonal antibody treatments |
| B.1.427/B.1.429 | 20C/S:452R | United States (June 2020) | L452R, W152C, S13I, D614G | Decrease in neutralization using convalescent and post-vaccination sera, ~20% increased transmissibility |
| P.2 | 20J | Brazil (April 2020) | L18F, T20N, P26S, F157L, E484K, D614G, S929I, V1176F | Potential depletion in neutralization by convalescent and post-vaccination sera or monoclonal antibody treatments |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens 2021, 10, 633. https://doi.org/10.3390/pathogens10060633
Janik E, Niemcewicz M, Podogrocki M, Majsterek I, Bijak M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens. 2021; 10(6):633. https://doi.org/10.3390/pathogens10060633
Chicago/Turabian StyleJanik, Edyta, Marcin Niemcewicz, Marcin Podogrocki, Ireneusz Majsterek, and Michal Bijak. 2021. "The Emerging Concern and Interest SARS-CoV-2 Variants" Pathogens 10, no. 6: 633. https://doi.org/10.3390/pathogens10060633
APA StyleJanik, E., Niemcewicz, M., Podogrocki, M., Majsterek, I., & Bijak, M. (2021). The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens, 10(6), 633. https://doi.org/10.3390/pathogens10060633