
Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Native LasB
2.2. Determination of Enzymatic Activity of nLasB
2.3. Phage Bio-Panning and Characterization of nLasB-Bound HuscFvs
2.4. Large Scale Production of Recombinant LasB-Bound HuscFvs
2.5. Fluorogenic Substrate Assay for Determining the LasB Neutralizing Activity of the HuscFvs
2.6. Elastin Congo Red Assay for Determining the HuscFvs-Mediated Inhibition of the LasB Elastolytic Activity
2.7. Homology Modeling and Inter-Molecular Docking
2.8. Statistical Analysis
3. Results
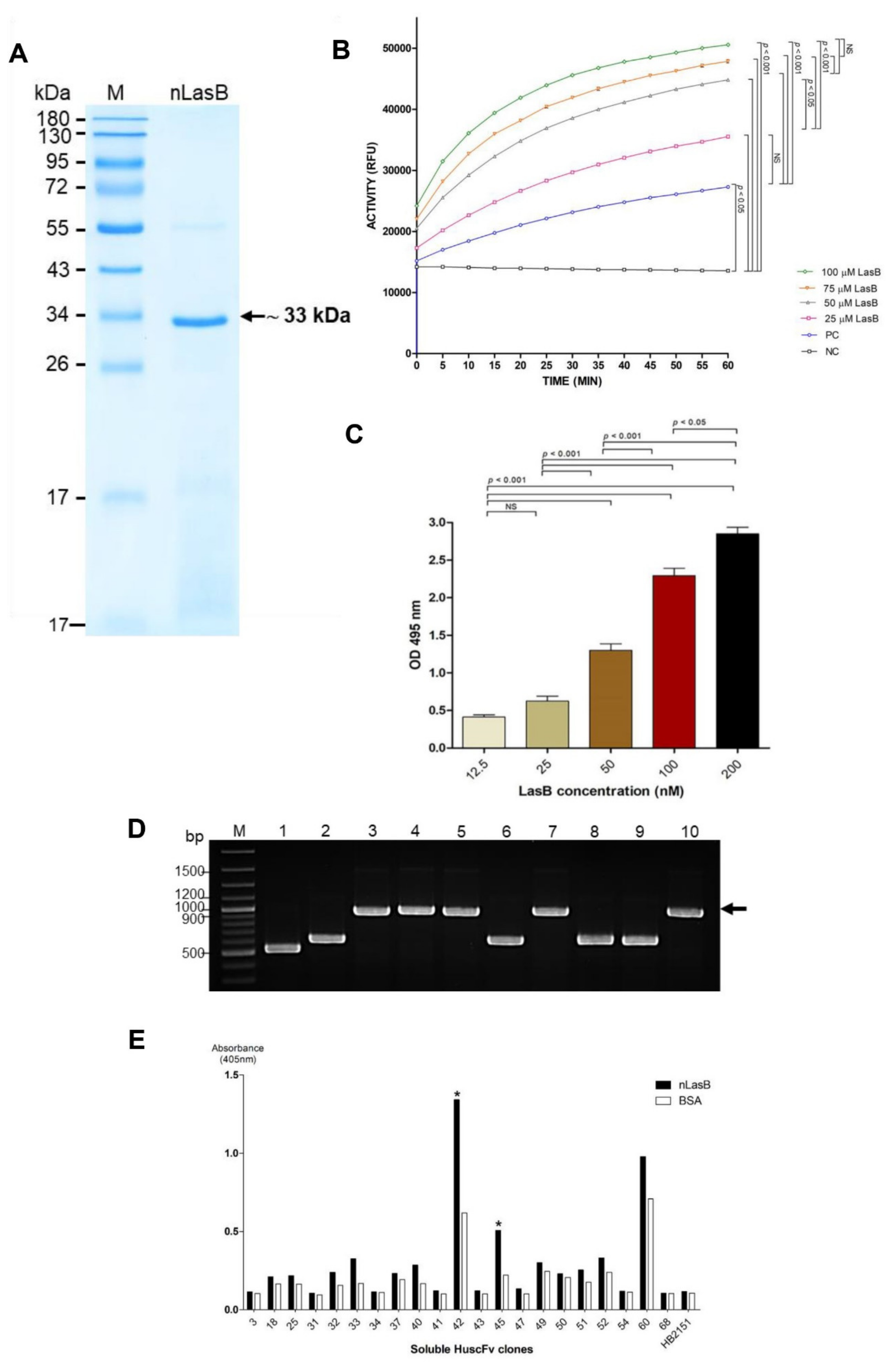
3.1. Purification and Characterization of the nLasB
3.2. Enzymatic Activities of the Native LasB
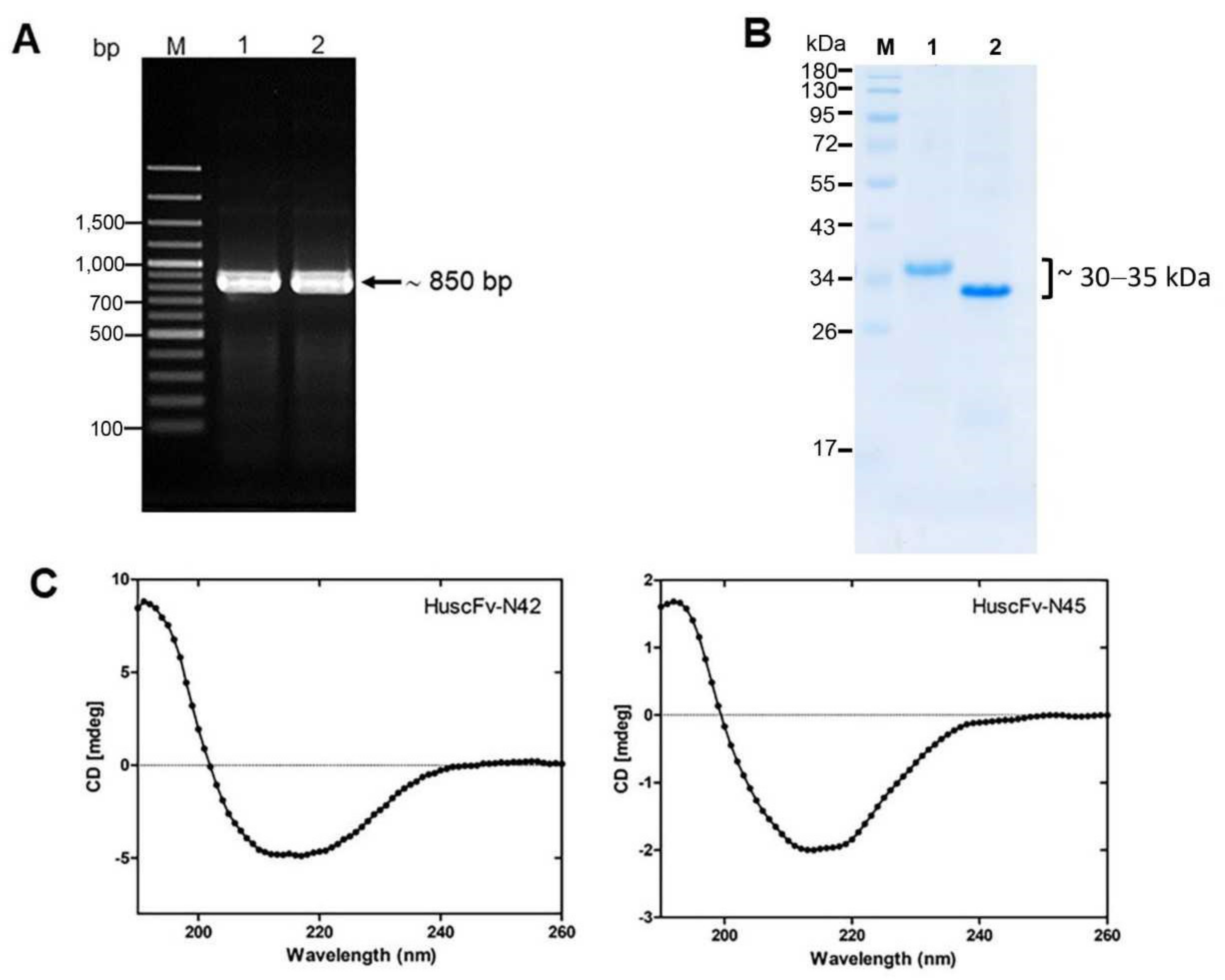
3.3. Selection and Characterization of LasB-Bound HuscFv-Displaying Phages
3.4. Recombinant HuscFvs to LasB
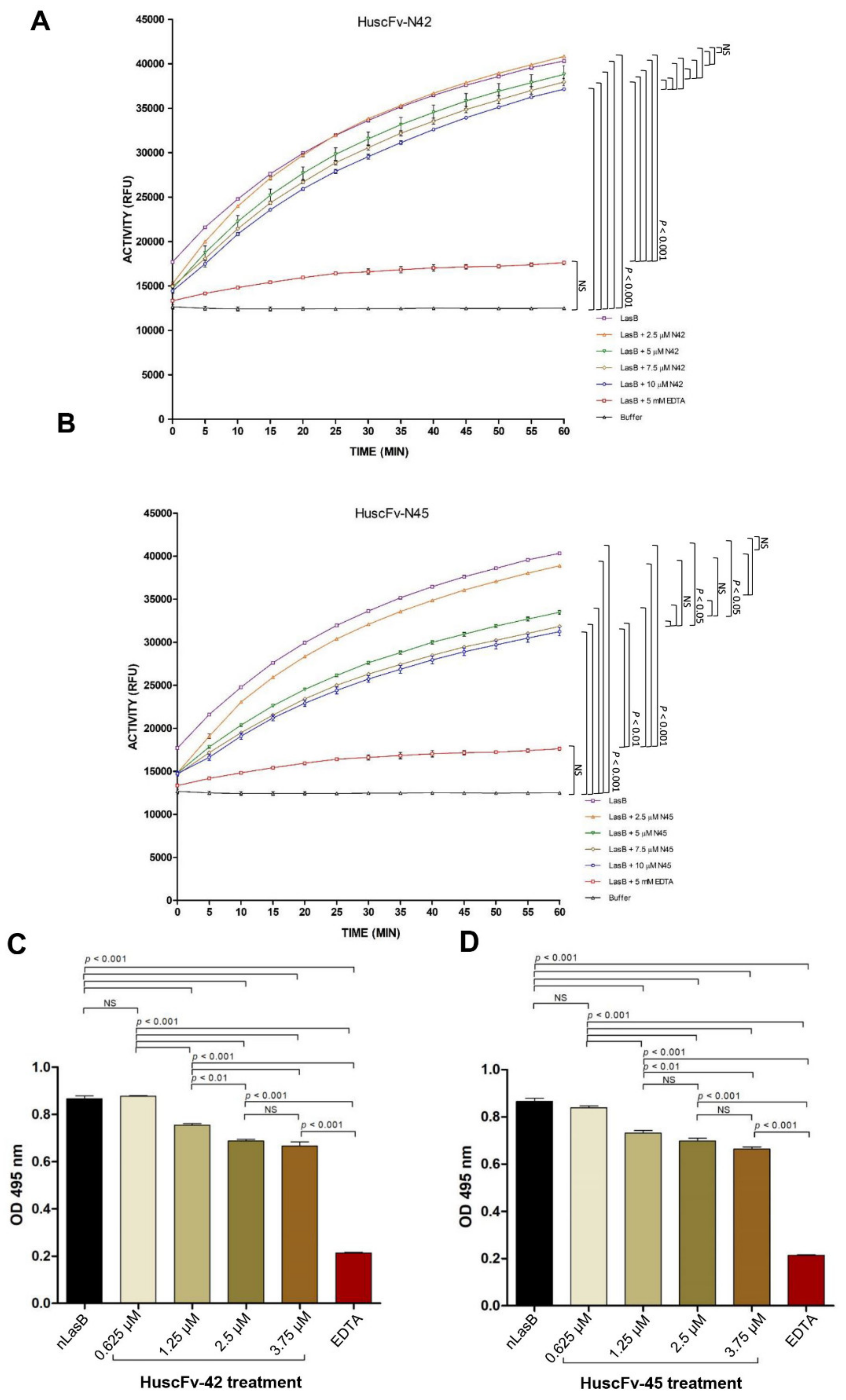
3.5. HuscFvs-Mediated Neutralization of the LasB Elastase Activity
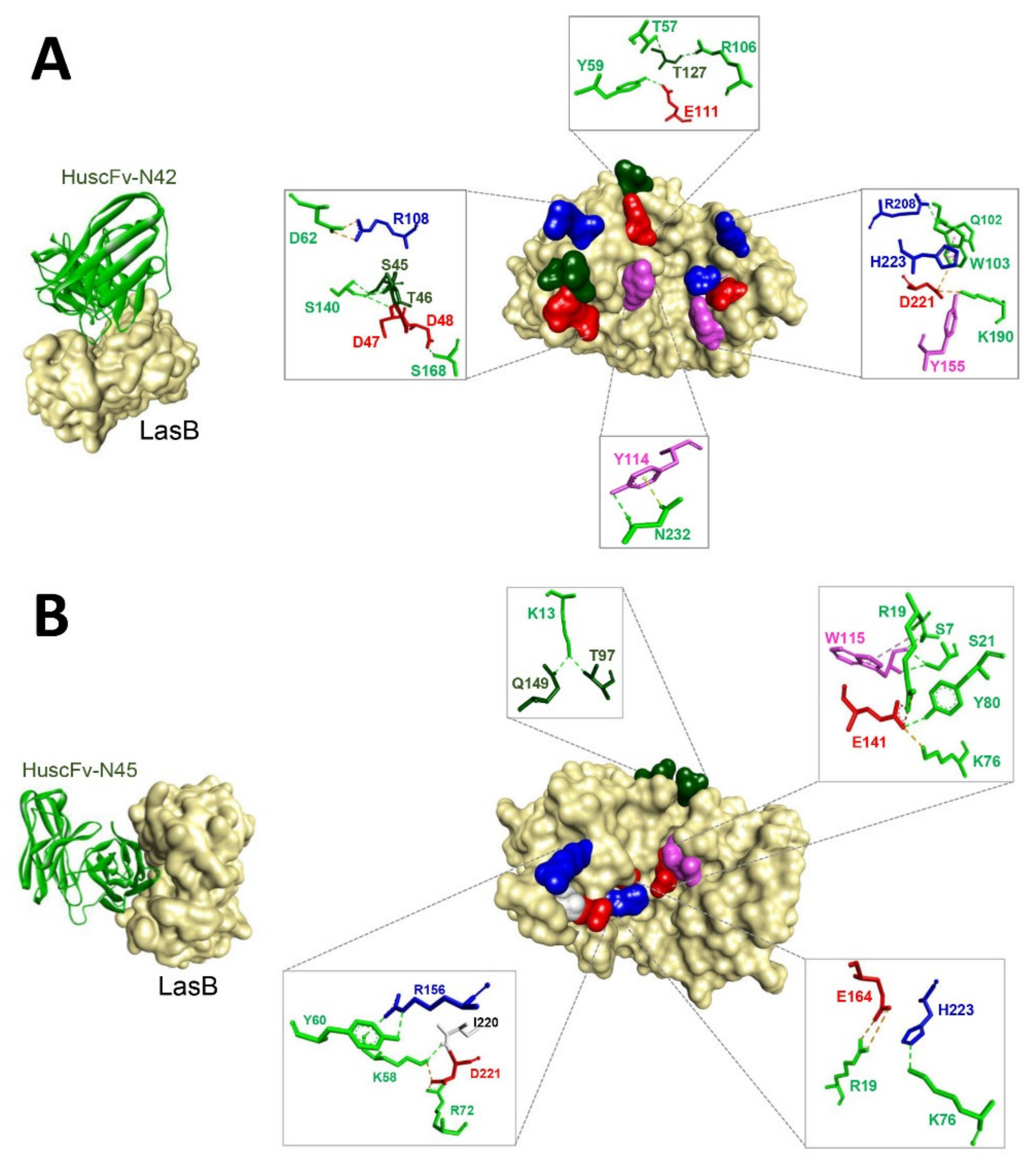
3.6. Predicted LasB Residues Bound by HuscFvs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morihara, K. Pseudolysin and other pathogen endopeptidases of thermolysin family. Methods Enzymol. 1995, 248, 242–253. [Google Scholar] [PubMed]
- Bever, R.A.; Iglewski, B.H. Molecular characterization and nucleotide sequence of the Pseudomonas aeruginosa elastase structural gene. J. Bacteriol. 1988, 170, 4309–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, P.; de Groot, A.; Bitter, W.; Tommassen, J. Secretion of elastinolytic enzymes and their propeptides by Pseudomonas aeruginosa. J. Bacteriol. 1998, 180, 3467–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, E.; Safrin, M.; Gustin, J.K.; Ohman, D.E. Elastase and the LasA protease of Pseudomonas aeruginosa are secreted with their propeptides. J. Biol. Chem. 1998, 273, 30225–30231. [Google Scholar] [CrossRef] [Green Version]
- Durand, É.; Bernadac, A.; Ball, G.; Lazdunski, A.; Sturgis, J.N.; Filloux, A. Type II protein secretion in Pseudomonas aeruginosa: The pseudopilus is a multifibrillar and adhesive structure. J. Bacteriol. 2003, 185, 2749–2758. [Google Scholar] [CrossRef] [Green Version]
- Filloux, A. Protein secretion systems in Pseudomonas aeruginosa: An essay on diversity, evolution, and function. Front. Microbiol. 2011, 2, 155. [Google Scholar] [CrossRef] [Green Version]
- Morihara, K.; Tsuzuki, H. Production of protease and elastase by Pseudomonas aeruginosa strains isolated from patients. Infect. Immun. 1977, 15, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef]
- Morihara, K.; Tsuzuki, H.; Oka, T.; Inoue, H.; Ebata, M. Pseudomonas aeruginosa elastase: Isolation, crystallization, and preliminary characterization. J. Biol. Chem. 1965, 240, 3295–3304. [Google Scholar] [CrossRef]
- Hersh, L.B.; Morihara, K. Comparison of the subsite specificity of the mammalian neutral endopeptidase 24.11 (enkephalinase) to the bacterial neutral endopeptidase thermolysin. J. Biol. Chem. 1986, 261, 6433–6437. [Google Scholar] [CrossRef]
- Galloway, D.R. Pseudomonas aeruginosa elastase and elastolysis revisited: Recent developments. Mol. Microbiol. 1991, 5, 2315–2321. [Google Scholar] [CrossRef]
- Heck, L.W.; Morihara, K.; McRae, W.B.; Miller, E.J. Specific cleavage of human type III and IV collagens by Pseudomonas aeruginosa elastase. Infect. Immun. 1986, 51, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Kessler, E.; Safrin, M. Elastinolytic and proteolytic enzymes. In Pseudomonas Methods and Protocols; Humana Press: New York, NY, USA, 2014; pp. 135–169. [Google Scholar]
- Döring, G.; Obernesser, H.J.; Botzenhart, K. Extracellular toxins of Pseudomonas aeruginosa. II. Effect of two proteases on human immunoglobulins IgG, IgA and secretory IgA. Zentralblatt Bakteriol. 1 Abt. Origin. A Med. Mikrobiol. Infekt. Parasitol. 1981, 249, 89–98. [Google Scholar]
- Holder, I.A.; Wheeler, R. Experimental studies of the pathogenesis of infections owing to Pseudomonas aeruginosa: Elastase, an IgG protease. Can. J. Microbiol. 1984, 30, 1118–1124. [Google Scholar] [CrossRef]
- Bastaert, F.; Kheir, S.; Saint-Criq, V.; Villeret, B.; Dang, P.M.C.; El-Benna, J.; Sirard, J.; Voulhoux, R.; Sallenave, J.M. Pseudomonas aeruginosa LasB subverts alveolar macrophage activity by interfering with bacterial killing through downregulation of innate immune defense, reactive oxygen species generation, and complement activation. Front. Immunol. 2018, 9, 1675. [Google Scholar] [CrossRef] [Green Version]
- Mariencheck, W.I.; Alcorn, J.F.; Palmer, S.M.; Wright, J.R. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am. J. Respir. Cell. Mol. Biol. 2003, 28, 528–537. [Google Scholar] [CrossRef]
- Komori, Y.; Nonogaki, T.; Nikai, T. Hemorrhagic activity and muscle damaging effect of Pseudomonas aeruginosa metalloproteinase (elastase). Toxicon 2001, 39, 1327–1332. [Google Scholar] [CrossRef]
- Azghani, A.O.; Miller, E.J.; Peterson, B.T. Virulence factors from Pseudomonas aeruginosa increase lung epithelial permeability. Lung 2000, 178, 261–269. [Google Scholar] [CrossRef]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef]
- Yanagihara, K.; Tomono, K.; Kaneko, Y.; Miyazaki, Y.; Tsukamoto, K.; Hirakata, Y.; Mukae, H. Role of elastase in a mouse model of chronic respiratory Pseudomonas aeruginosa infection that mimics diffuse panbronchiolitis. J. Med. Microbiol. 2003, 52, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Schmidtchen, A.; Holst, E.; Tapper, H.; Björck, L. Elastase-producing Pseudomonas aeruginosa degrade plasma proteins and extracellular products of human skin and fibroblasts, and inhibit fibroblast growth. Microb. Pathog. 2003, 34, 47–55. [Google Scholar] [CrossRef]
- Hobden, J.A. Pseudomonas aeruginosa proteases and corneal virulence. DNA Cell Biol. 2002, 21, 391–396. [Google Scholar] [CrossRef]
- Cathcart, G.R.; Quinn, D.; Greer, B.; Harriott, P.; Lynas, J.F.; Gilmore, B.F.; Walker, B. Novel inhibitors of the Pseudomonas aeruginosa virulence factor LasB: A potential therapeutic approach for the attenuation of virulence mechanisms in pseudomonal infection. Antimicrob. Agents Chemother. 2011, 55, 2670–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Cai, X.; Harris, T.L.; Gooyit, M.; Wood, M.; Lardy, M.; Janda, K.D. Disarming Pseudomonas aeruginosa virulence factor LasB by leveraging a Caenorhabditis elegans infection model. Chem. Biol. 2015, 22, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; LaRock, D.; Skowronski, E.; Kimmey, J.M.; Olson, J.; Jiang, Z.; O′Donoghue, A.J. Role of inflammasome-independent activation of IL-1β by the Pseudomonas aeruginosa protease LasB. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cowell, B.A.; Twining, S.S.; Hobden, J.A.; Kwong, M.S.; Fleiszig, S.M. Mutation of lasA and lasB reduces Pseudomonas aeruginosa invasion of epithelial cells. Microbiology 2003, 149, 2291–2299. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.B.; DiMango, E.; Bryan, R.; Gambello, M.; Iglewski, B.H.; Goldberg, J.B.; Prince, A. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect. Immun. 1996, 64, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.W.; Rahme, L.G.; Sternberg, J.A.; Tompkins, R.G.; Ausubel, F.M. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. USA 1999, 96, 2408–2413. [Google Scholar] [CrossRef] [Green Version]
- Schimmel, P.; Tao, J.; Hill, J. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 1998, 12, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Bryskier, A. Antimicrobial Agents: Antibacterials and Antifungals; ASM Press: Washington, DC, USA, 2005; ISBN 1-55581-237-6. [Google Scholar]
- Hu, Y.; Keniry, M.; Palmer, S.O.; Bullard, J.M. Discovery and analysis of natural-product compounds inhibiting protein synthesis in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 4820. [Google Scholar] [CrossRef] [Green Version]
- Galdino, A.C.M.; Viganor, L.; de Castro, A.A.; da Cunha, E.F.; Mello, T.P.; Mattos, L.M.; Pereira, M.D.; Hunt, M.C.; O′Shaughnessy, M.; Howe, O.; et al. Disarming Pseudomonas aeruginosa virulence by the inhibitory action of 1, 10-phenanthroline-5, 6-dione-based compounds: Elastase B (lasB) as a chemotherapeutic target. Front. Microbiol. 2019, 10, 1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adekoya, O.A.; Sjøli, S.; Wuxiuer, Y.; Bilto, I.; Marques, S.M.; Santos, M.A.; Nuti, E.; Cercignani, G.; Rossello, A.; Winberg, J.O.; et al. Inhibition of pseudolysin and thermolysin by hydroxamate-based MMP inhibitors. Eur. J. Med. Chem. 2015, 89, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Herrington-Symes, A.P.; Farys, M.; Khalili, H.; Brocchini, S. Antibody fragments: Prolonging circulation half-life special issue-antibody research. Adv. Biosci. Biotechnol. 2013, 4, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteom. 2009, 72, 270–282. [Google Scholar] [CrossRef]
- Chaisri, U.; Chaicumpa, W. Evolution of therapeutic antibodies, influenza virus biology, influenza, and influenza immunotherapy. BioMed Res. Int. 2018, 2018, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Abhinandan, K.R.; Martin, A.C. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol. Immunol. 2008, 45, 3832–3839. [Google Scholar] [CrossRef]
- Santajit, S.; Seesuay, W.; Mahasongkram, K.; Sookrung, N.; Pumirat, P.; Ampawong, S.; Reamtong, O.; Chongsa-Nguan, M.; Chaicumpa, W.; Indrawattana, N. Human single-chain variable fragments neutralize Pseudomonas aeruginosa quorum sensing molecule, 3O-C12-HSL, and prevent cells from the HSL-mediated apoptosis. Front. Microbiol. 2020, 11, 1172. [Google Scholar] [CrossRef]
- Thayer, M.M.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-Å resolution. J. Biol. Chem. 1991, 266, 2864–2871. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nature Prot. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [Green Version]
- Brenke, R.; Hall, D.R.; Chuang, G.Y.; Comeau, S.R.; Bohnuud, T.; Beglov, D.; Schueler-Furman, O.; Vajda, S.; Kozakov, D. Application of asymmetric statistical potentials to antibody–protein docking. Bioinformatics 2012, 28, 2608–2614. [Google Scholar] [CrossRef] [Green Version]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein–protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nature Protoc. 2017, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Kessler, E.; Safrin, M. Synthesis, processing, and transport of Pseudomonas aeruginosa elastase. J. Bacteriol. 1988, 170, 5241–5247. [Google Scholar] [CrossRef] [Green Version]
- Kessler, E.; Safrin, M.; Peretz, M.; Burstein, Y. Identification of cleavage sites involved in proteolytic processing of Pseudomonas aeruginosa preproelastase. FEBS Lett. 1992, 299, 291–293. [Google Scholar] [CrossRef] [Green Version]
- McIver, K.S.; Kessler, E.; Ohman, D.E. Substitution of active-site His-223 in Pseudomonas aeruginosa elastase and expression of the mutated lasB alleles in Escherichia coli show evidence for autoproteolytic processing of proelastase. J. Bacteriol. 1991, 173, 7781–7789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIver, K.S.; Kessler, E.; Olson, J.C.; Ohman, D.E. The elastase propeptide functions as an intramolecular chaperone required for elastase activity and secretion in Pseudomonas aeruginosa. Mol. Microbiol. 1995, 18, 877–889. [Google Scholar] [CrossRef]
- Braun, P.; Tommassen, J.; Filloux, A. Role of the propeptide in folding and secretion of elastase of Pseudomonas aeruginosa. Mol. Microbiol. 1996, 19, 297–306. [Google Scholar] [CrossRef]
- Cathcart, G.; Gilmore, B.; Walker, B. Enzyme Kinetic Measurements for a Combinatorial Library of Inhibitors of Pseudomonas Elastase. School of Pharmacy, QUB, Belfast. BMG LABTECH. Available online: https://pure.qub.ac.uk. (accessed on 10 June 2021).
- Padlan, E. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol. Immunol. 1991, 28, 489–498. [Google Scholar] [CrossRef]
- Shi, B.; Ma, L.; He, X.; Wang, X.; Wang, P.; Zhou, L.; Yao, X. Comparative analysis of human and mouse immunoglobulin variable heavy regions from IMGT/LIGM-DB with IMGT/HighV-QUEST. Theor. Biol. Med. Model. 2014, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Salvesen, G. Handbook of Proteolytic Enzymes, 3rd ed.; Academic Press: Amsterdam, The Netherlands, 2013; Volume 3. [Google Scholar]
- Campa, M.; Bendinelli, M.; Friedman, H. Pseudomonas Aeruginosa As an Opportunistic Pathogen; Springer Science & Business Media: New York, NY, USA, 2012. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LasB Protein | HuscFv-N42 | Interactive Bond (s) | |
|---|---|---|---|
| Residue | Residue | Domain (s) | |
| S45 | S140 | Linker | Hydrogen |
| T46 | S140 | Linker | Hydrogen |
| D47 | S140 | Linker | Hydrogen |
| D48 | S168 | VL-CDR1 | Hydrogen |
| R108 | D62 | VH-CDR2 | Salt bridge |
| E111 | Y59 | VH-CDR2 | Hydrogen |
| Y114 | N232 | VL-CDR3 | Hydrogen |
| T127 | T57/R106 | VH-CDR2/CDR3 | Hydrogen |
| Y155 (substrate binding) | K190 | VL-CDR2 | Hydrogen |
| R208 | Q102 | VH-CDR3 | Hydrogen |
| D221 (substrate binding) | K190 | VL-CDR2 | Salt bridge |
| H223 (substrate binding) | W103 | VH-CDR3 | Hydrophobic (π-π stacking) |
| LasB Protein | HuscFv-N45 | Interactive Bond(s) | |
| Residue | Residue | Domain(s) | |
| T97 | K13 | VH-FR1 | Hydrogen |
| W115 | S7/R19/S21 | VH-FR1 | Hydrogen |
| E141 (located at the center of the catalytic site) | K76/Y80 | VH-FR1/FR3 | Salt bridge |
| Q149 | K13 | VH-FR1 | Hydrogen |
| R156 | K58/Y60 | VH-CDR2 | Hydrogen |
| E164 (ligand of zinc co-factor) | R19 | VH-FR1 | Salt Bridge |
| I220 | K58 | VH-CDR2 | Hydrogen |
| D221 (substrate binding) | K58/R72 | VH-CDR2/FR3 | Ionic |
| H223 (substrate binding) | K76 | VH-FR3 | Hydrogen |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santajit, S.; Kong-ngoen, T.; Chongsa-Nguan, M.; Boonyuen, U.; Pumirat, P.; Sookrung, N.; Chaicumpa, W.; Indrawattana, N. Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB. Pathogens 2021, 10, 765. https://doi.org/10.3390/pathogens10060765
Santajit S, Kong-ngoen T, Chongsa-Nguan M, Boonyuen U, Pumirat P, Sookrung N, Chaicumpa W, Indrawattana N. Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB. Pathogens. 2021; 10(6):765. https://doi.org/10.3390/pathogens10060765
Chicago/Turabian StyleSantajit, Sirijan, Thida Kong-ngoen, Manas Chongsa-Nguan, Usa Boonyuen, Pornpan Pumirat, Nitat Sookrung, Wanpen Chaicumpa, and Nitaya Indrawattana. 2021. "Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB" Pathogens 10, no. 6: 765. https://doi.org/10.3390/pathogens10060765
APA StyleSantajit, S., Kong-ngoen, T., Chongsa-Nguan, M., Boonyuen, U., Pumirat, P., Sookrung, N., Chaicumpa, W., & Indrawattana, N. (2021). Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB. Pathogens, 10(6), 765. https://doi.org/10.3390/pathogens10060765