
Molecular Characterization of the env Gene of Bovine Leukemia Virus in Cattle from Pakistan with NGS-Based Evidence of Virus Heterogeneity

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Results of PCR Amplification on Blood and Tissue Samples
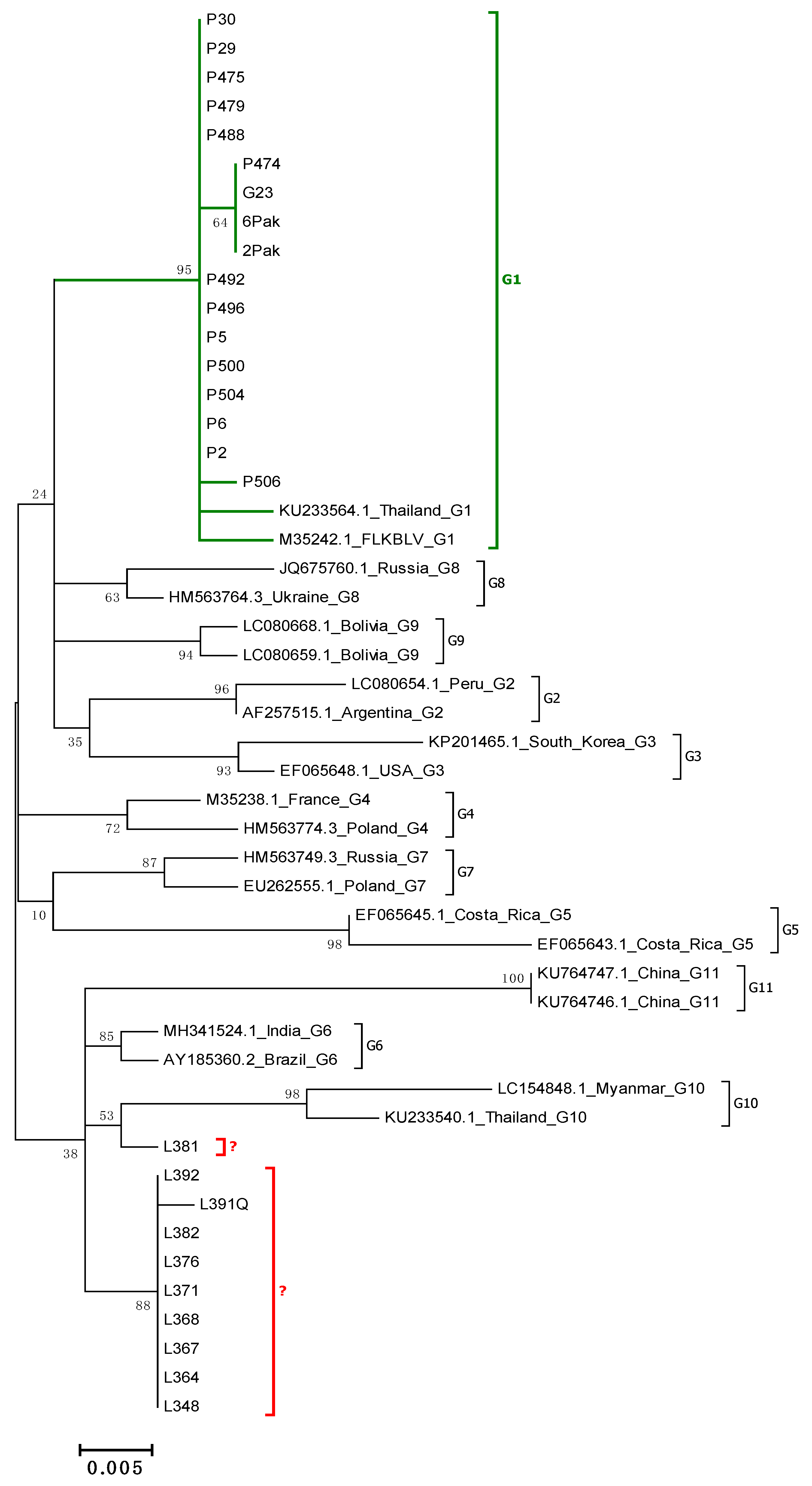
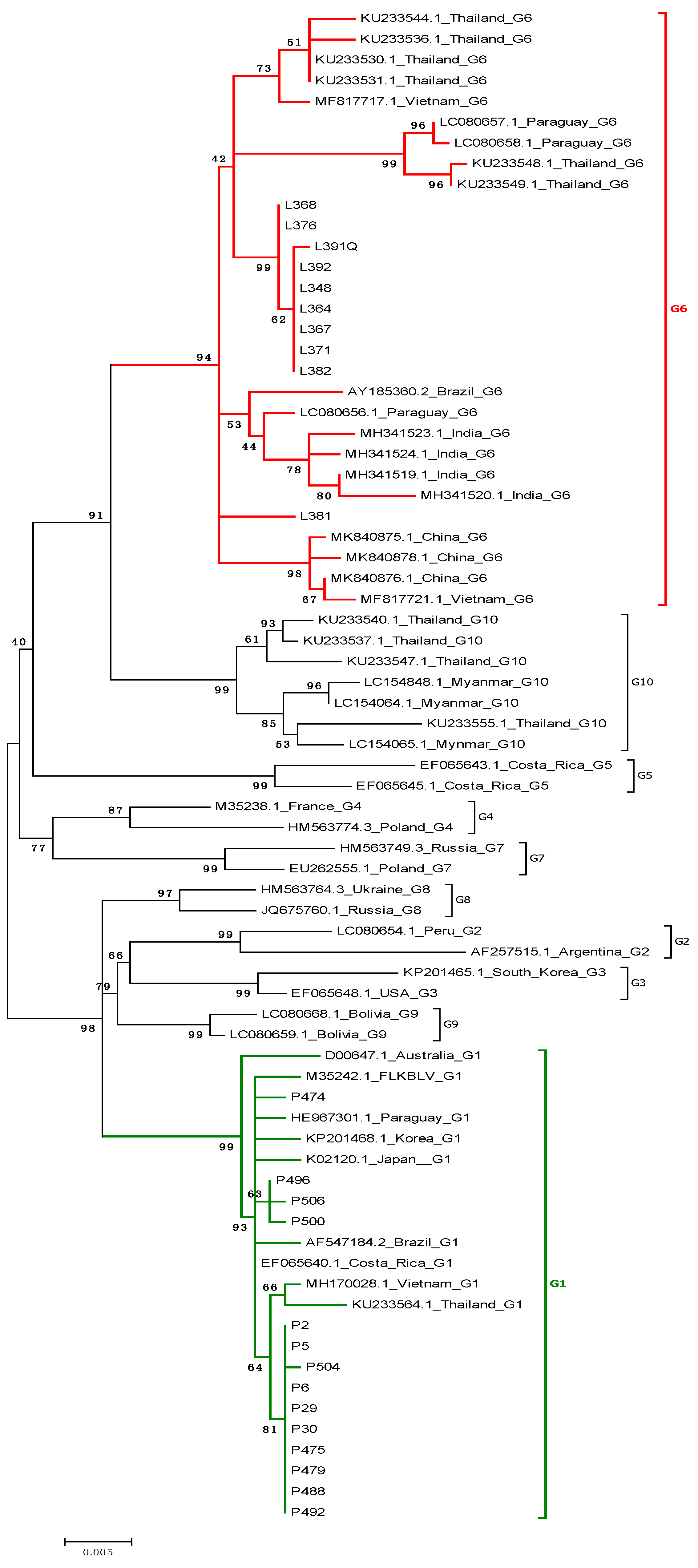
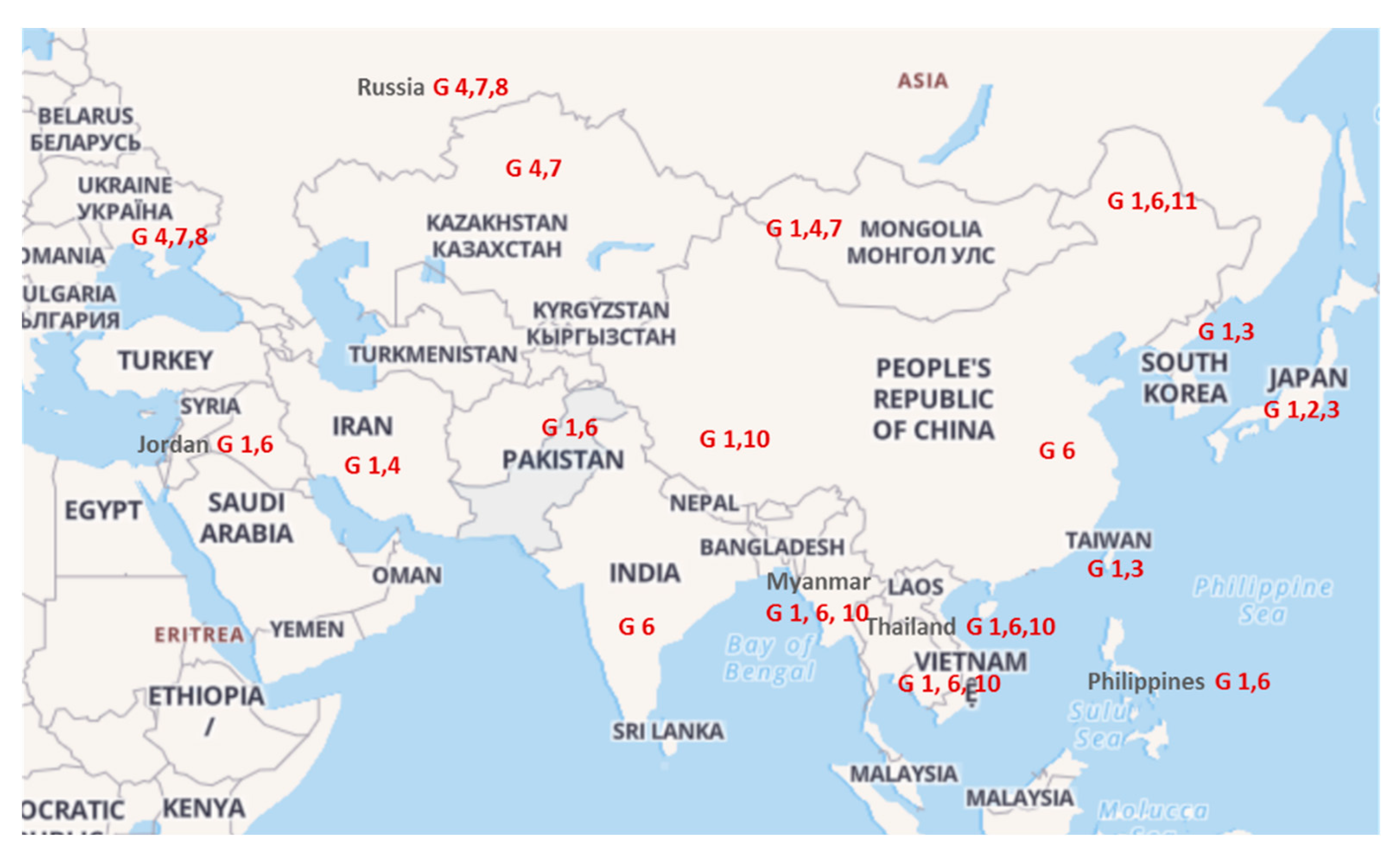
2.2. Phylogenetic Analysis Based on 400 and 903 bp env Gene Sequences
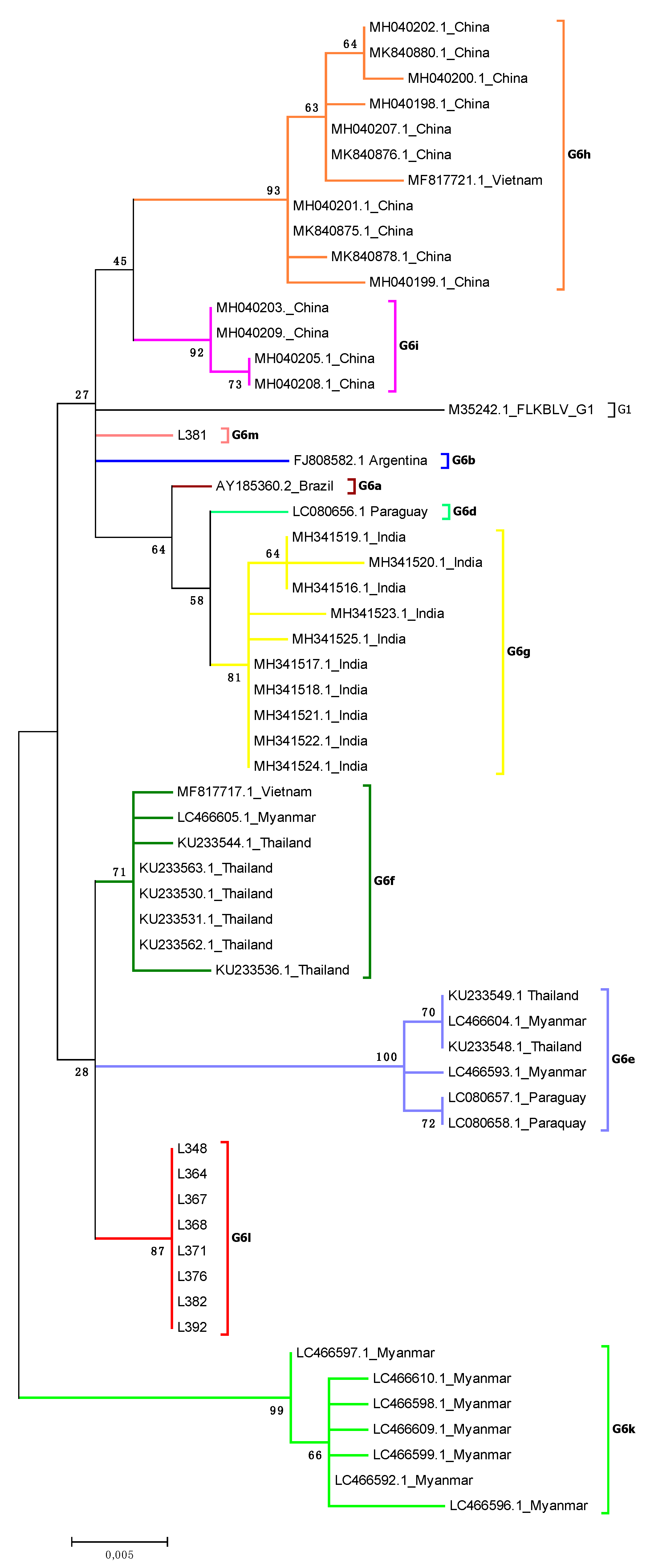
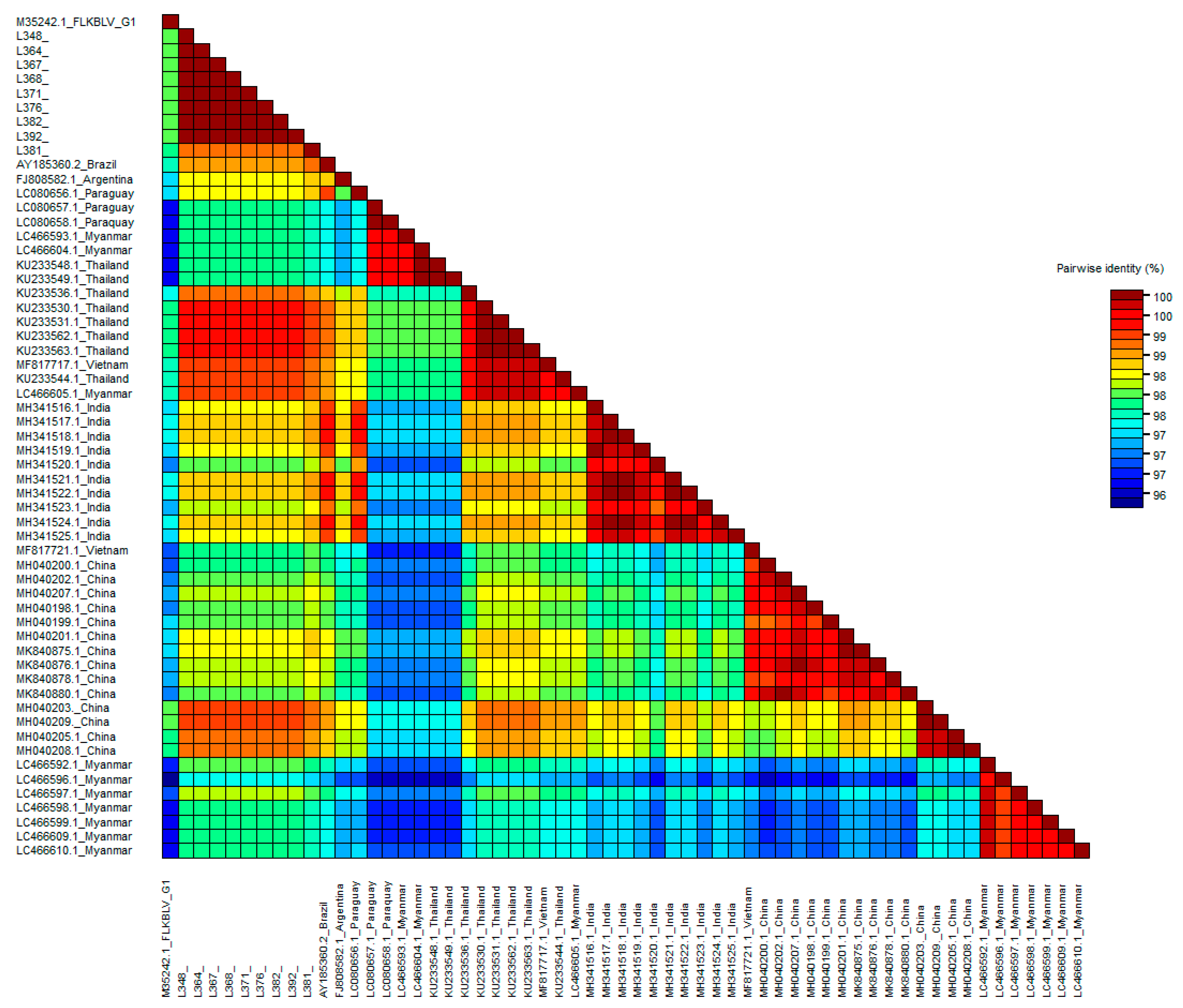
2.3. Analysis of Subtyping within Genotype G6
2.4. Comparison of Nucleotide Sequences Belonging to Genotype G1 and G6
2.5. Nucleotide and Amino Acid Sequence Analysis
2.6. NGS Analysis of DNA from Sample L391
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. PCR Amplification of 444 bp and 993 bp Fragments of env Gene
4.3. DNA Sequencing and Sequence Analysis
4.4. Next-Generation Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gillet, N.; Florins, A.; Boxus, M.; Burteau, C.; Nigro, A.; Vandermeers, F.; Balon, H.; Bouzar, A.-B.; Defoiche, J.; Burny, A.; et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: Prospects for novel anti-retroviral therapies in human. Retrovirology 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, Y.; Murakami, H.; Takahashi, M.; Takeshima, S.-N. Mechanisms of pathogenesis induced by bovine leukemia virus as a model for human T-cell leukemia virus. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, I.; Lévy, D. Pathobiology of bovine leukemia virus. Vet. Res. 1994, 25, 521–536. [Google Scholar] [PubMed]
- Polat, M.; Takeshima, S.; Aida, Y. Epidemiology and genetic diversity of bovine leukemia virus. Virol. J. 2017, 14. [Google Scholar] [CrossRef]
- Taylor, D.B. EFSA AHAW Panel (EFSA Panel on Animal Health and Welfare) Scientific Opinion on enzootic bovine leukosis. EFSA J. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, P.C.; Norby, B.; Byrem, T.M.; Parmelee, A.; Ledergerber, J.T.; Erskine, R.J. Bovine leukemia virus and cow longevity in Michigan dairy herds. J. Dairy Sci. 2013, 96, 1591–1597. [Google Scholar] [CrossRef] [Green Version]
- Ott, S.L.; Johnson, R.; Wells, S.J. Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev. Vet. Med. 2003, 61, 249–262. [Google Scholar] [CrossRef]
- Erskine, R.J.; Bartlett, P.C.; Byrem, T.M.; Render, C.L.; Febvay, C.; Houseman, J.T. Association between bovine leukemia virus, production, and population age in Michigan dairy herds. J. Dairy Sci. 2012, 95, 727–734. [Google Scholar] [CrossRef]
- Tsutsui, T.; Kobayashi, S.; Hayama, Y.; Nishiguchi, A.; Kameyama, K.; Konishi, M.; Murakami, K. Estimation of the within-herd transmission parameter of bovine leukemia virus. Prev. Vet. Med. 2010, 95. [Google Scholar] [CrossRef]
- Blagitz, M.G.; Souza, F.N.; Batista, C.F.; Azevedo, L.F.F.; Sanchez, E.M.R.; Diniz, S.A.; Silva, M.X.; Haddad, J.P.; Della Libera, A.M.M.P. Immunological implications of bovine leukemia virus infection. Res. Vet. Sci. 2017, 114. [Google Scholar] [CrossRef]
- Chi, J.; VanLeeuwen, J.A.; Weersink, A.; Keefe, G.P. Management factors related to seroprevalences to bovine viral-diarrhoea virus, bovine-leukosis virus, Mycobacterium avium subspecies paratuberculosis, and Neospora caninum in dairy herds in the Canadian Maritimes. Prev. Vet. Med. 2002, 55. [Google Scholar] [CrossRef]
- Hopkins, S.G.; DiGiacomo, R.F. Natural transmission of bovine leukemia virus in dairy and beef cattle. Vet. Clin. North Am. Food Anim. Pract. 1997, 13, 107–128. [Google Scholar] [CrossRef]
- Meas, S.; Usui, T.; Ohashi, K.; Sugimoto, C.; Onuma, M. Vertical transmission of bovine leukemia virus and bovine immunodeficiency virus in dairy cattle herds. Vet. Microbiol. 2002, 84, 275–282. [Google Scholar] [CrossRef]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, E.R.; Albritton, L.M.; Radke, K. Envelope Proteins Containing Single Amino Acid Substitutions Support a Structural Model of the Receptor-Binding Domain of Bovine Leukemia Virus Surface Protein. J. Virol. 2002, 76, 10861–10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavanya, M.; Kinet, S.; Montel-Hagen, A.; Mongellaz, C.; Battini, J.-L.; Sitbon, M.; Taylor, N. Cell Surface Expression of the Bovine Leukemia Virus-Binding Receptor on B and T Lymphocytes Is Induced by Receptor Engagement. J. Immunol. 2008, 181, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Bruck, C.; Portetelle, D.; Burny, A.; Zavada, J. Topographical analysis by monoclonal antibodies of BLV-gp51 epitopes involved in viral functions. Virology 1982, 122, 353–362. [Google Scholar] [CrossRef]
- Bruck, C.; Rensonnet, N.; Portetelle, D.; Cleuter, Y.; Mammerickx, M.; Burny, A.; Mamoun, R.; Guillemain, B.; Van Der Maaten, M.J.; Ghysdael, J. Biologically active epitopes of bovine leukemia virus glycoprotein GP51: Their dependence on protein glycosylation and genetic variability. Virology 1984, 136, 20–31. [Google Scholar] [CrossRef]
- Callebaut, I.; Vonèche, V.; Mager, A.; Fumière, O.; Krchnak, V.; Merza, M.; Zavada, J.; Mammerickx, M.; Burny, A.; Portetelle, D. Mapping of B-neutralizing and T-helper cell epitopes on the bovine leukemia virus external glycoprotein gp51. J. Virol. 1993, 67, 5321–5327. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Takeshima, S.; Isogai, E.; Kohara, J.; Aida, Y. Novel CD8+ cytotoxic T cell epitopes in bovine leukemia virus with cattle. Vaccine 2015, 33, 7194–7202. [Google Scholar] [CrossRef] [Green Version]
- Gatot, J.-S.; Callebaut, I.; Van Lint, C.; Demonté, D.; Kerkhofs, P.; Portetelle, D.; Burny, A.; Willems, L.; Kettmann, R. Bovine Leukemia Virus SU Protein Interacts with Zinc, and Mutations within Two Interacting Regions Differently Affect Viral Fusion and Infectivity In Vivo. J. Virol. 2002, 76, 7956–7967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moratorio, G.; Obal, G.; Dubra, A.; Correa, A.; Bianchi, S.; Buschiazzo, A.; Cristina, J.; Pritsch, O. Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch. Virol. 2010, 155, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.M.; Golemba, M.D.; Campos, R.H.; Trono, K.; Jones, L.R. Bovine leukemia virus can be classified into seven genotypes: Evidence for the existence of two novel clades. J. Gen. Virol. 2009, 90, 2788–2797. [Google Scholar] [CrossRef]
- Camargos, M.F.; Pereda, A.; Stancek, D.; Rocha, M.A.; dos Reis, J.K.P.; Greiser-Wilke, I.; Leite, R.C. Molecular characterization of the env gene from Brazilian field isolates of Bovine Leukemia Virus. Virus Genes 2007, 34, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, E.-J.; Joung, H.-K.; Kim, B.-H.; Song, J.-Y.; Cho, I.-S.; Lee, K.-K.; Shin, Y.-K. Sequencing and phylogenetic analysis of the gp51 gene from Korean bovine leukemia virus isolates. Virol. J. 2015, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- Rola-Łuszczak, M.; Pluta, A.; Olech, M.; Donnik, I.; Petropavlovskiy, M. The Molecular Characterization of Bovine Leukaemia Virus Isolates from Eastern Europe and Siberia and Its Impact on Phylogeny. PLoS ONE 2013, 8, 58705. [Google Scholar] [CrossRef]
- Lee, E.; Kim, E.-J.; Ratthanophart, J.; Vitoonpong, R.; Kim, B.-H.; Cho, I.-S.; Song, J.-Y.; Lee, K.-K.; Shin, Y.-K. Molecular epidemiological and serological studies of bovine leukemia virus (BLV) infection in Thailand cattle. Infect. Genet. Evol. 2016, 41, 245–254. [Google Scholar] [CrossRef]
- Polat, M.; Moe, H.H.; Shimogiri, T.; Moe, K.K.; Takeshima, S.; Aida, Y. The molecular epidemiological study of bovine leukemia virus infection in Myanmar cattle. Arch. Virol. 2017, 162, 425–437. [Google Scholar] [CrossRef]
- Suzuki, A.; Chapman, R.; Douglass, N.; Carulei, O.; van Rensburg, J.; Williamson, A.-L. Phylogenetic Analysis of South African Bovine Leukaemia Virus (BLV) Isolates. Viruses 2020, 12, 898. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Sood, R. Molecular Characterization of Bovine Leukaemia Virus (BLV) Strains Reveals Existence of Genotype 6 in Cattle in India with evidence of a new subgenotype. Transbound. Emerg. Dis. 2018, 65, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Hemmatzadeh, F. Sequencing and Phylogenetic Analysis of gp51 Gene of Bovine Leukaemia Virus in Iranian Isolates. Vet. Res. Commun. 2007, 31, 783–789. [Google Scholar] [CrossRef]
- Felmer, R.; Muñoz, G.; Zúñiga, J.; Recabal, M. Molecular analysis of a 444bp fragment of the bovine leukaemia virus gp51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet. Microbiol. 2005, 108, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Balić, D.; Lojkić, I.; Periškić, M.; Bedeković, T.; Jungić, A.; Lemo, N.; Roić, B.; Čač, Ž.; Barbić, L.; Madić, J. Identification of a new genotype of bovine leukemia virus. Arch. Virol. 2012, 157, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Polat, M.; Takeshima, S.; Hosomichi, K.; Kim, J.; Miyasaka, T.; Yamada, K.; Arainga, M.; Murakami, T.; Matsumoto, Y.; de la Barra Diaz, V.; et al. A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology 2016, 13, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Wang, X.; Zhou, Y.; Wang, Y.; Zhang, X.; Zheng, Y. Genotyping bovine leukemia virus in dairy cattle of Heilongjiang, northeastern China. BMC Vet. Res. 2019, 15, 179. [Google Scholar] [CrossRef]
- de Brogniez, A.; Bouzar, A.B.; Jacques, J.-R.; Cosse, J.-P.; Gillet, N.; Callebaut, I.; Reichert, M.; Willems, L. Mutation of a Single Envelope N-Linked Glycosylation Site Enhances the Pathogenicity of Bovine Leukemia Virus. J. Virol. 2015, 89, 8945–8956. [Google Scholar] [CrossRef] [Green Version]
- de Brogniez, A.; Mast, J.; Willems, L. Determinants of the Bovine Leukemia Virus Envelope Glycoproteins Involved in Infectivity, Replication and Pathogenesis. Viruses 2016, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Fechner, H.; Blankenstein, P.; Looman, A.C.; Elwert, J.; Geue, L.; Albrecht, C.; Kurg, A.; Beier, D.; Marquardt, O.; Ebner, D. Provirus Variants of the Bovine Leukemia Virus and Their Relation to the Serological Status of Naturally Infected Cattle. Virology 1997, 237, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Pakistan Economic Survey 2019–2020. Available online: http://www.finance.gov.pk/survey/chapter_20/PES_2019_20.pdf (accessed on 10 June 2021).
- Khan, M.F.; Siddique, U.; Shah, A.A.; Khan, I.; Anwar, F.; Ahmad, I.; Zeb, M.T.; Hassan, M.F.; Ali, T. Seroprevalence of bovine leukemia virus (BLV) in cattle from the North West of Pakistan. Pak. Vet. J. 2020, 40, 127–129. [Google Scholar] [CrossRef]
- Sakhawat, A.; Rola-Łuszczak, M.; Osiński, Z.; Bibi, N.; Kuźmak, J. Bayesian Estimation of the True Seroprevalence and Risk Factor Analysis of Bovine Leukemia Virus Infection in Pakistan. Animals 2021, 11, 1404. [Google Scholar] [CrossRef]
- Dao, T.D.; Bui, V.N.; Omatsu, T.; Katayama, Y.; Mizutani, T.; Ogawa, H.; Imai, K. Application of the SureSelect target enrichment system for next-generation sequencing to obtain the complete genome sequence of bovine leukemia virus. Arch. Virol. 2018, 163, 3155–3159. [Google Scholar] [CrossRef]
- Moe, K.K.; Polat, M.; Borjigin, L.; Matsuura, R.; Hein, S.T.; Moe, H.H.; Aida, Y. New evidence of bovine leukemia virus circulating in Myanmar cattle through epidemiological and molecular characterization. PLoS ONE 2020, 15, e0229126. [Google Scholar] [CrossRef] [Green Version]
- Hamada, R.; Metwally, S.; Polat, M.; Borjigin, L.; Ali, A.O.; Abdel-Hady, A.A.A.; Mohamed, A.E.A.; Wada, S.; Aida, Y. Detection and Molecular Characterization of Bovine Leukemia Virus in Egyptian Dairy Cattle. Front. Vet. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Ochirkhuu, N.; Konnai, S.; Odbileg, R.; Nishimori, A.; Okagawa, T.; Murata, S.; Ohashi, K. Detection of bovine leukemia virus and identification of its genotype in Mongolian cattle. Arch. Virol. 2016, 161, 985–991. [Google Scholar] [CrossRef]
- Vafin, R.R.; Khazipov, N.Z.; Shaeva, A.I.; Zakirova, Z.R.; Zaĭnullin, L.I.; Tiul’kin, S.V.; Abdulina, I.R.; Alimov, A.M. Genotypic identification of the bovine leukemia virus. Mol. Gen. Mikrobiol. Virusol. 2014, 4, 34–40. [Google Scholar] [CrossRef]
- Baszenova, E.; Mamonova, S.; Saduakassova, M.; Sultanov, A.; (KazSRVI Almaty, Republic of Kazakhstan); Rola-Łuszczak, M.; Ryło, A.; Osiński, Z.; Kuźmak, J.; (NVRI Pulawy, Poland). Personal Communication, 2021. Unpublished work.
- Yang, Y.; Chu, S.; Shang, S.; Yang, Z.; Wang, C. Short communication: Genotyping and single nucleotide polymorphism analysis of bovine leukemia virus in Chinese dairy cattle. J. Dairy Sci. 2019, 102, 3469–3473. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, L.; Dong, M.; Huang, W.; Hao, X.; Peng, Y.; Gong, Z.; Qin, A.; Shang, S.; Yang, Z. Molecular characterization of bovine leukemia virus reveals existence of genotype 4 in Chinese dairy cattle. Virol. J. 2019, 16, 108. [Google Scholar] [CrossRef] [Green Version]
- Ababneh, M.M.; Al-Rukibat, R.K.; Hananeh, W.M.; Nasar, A.T.; Al-Zghoul, M.B. Detection and molecular characterization of bovine leukemia viruses from Jordan. Arch. Virol. 2012, 157, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Yamashita-Kawanishi, N.; Okamoto, M.; Nguyen, S.V.; Nguyen, N.H.; Sugiura, K.; Miura, T.; Haga, T. Detection and genotyping of bovine leukemia virus (Blv) in vietnamese cattle. J. Vet. Med. Sci. 2020, 82, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Polat, M.; Ohno, A.; Takeshima, S.; Kim, J.; Kikuya, M.; Matsumoto, Y.; Mingala, C.N.; Onuma, M.; Aida, Y. Detection and molecular characterization of bovine leukemia virus in Philippine cattle. Arch. Virol. 2015, 160, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.-C.; Li, C.-Y.; Hsu, W.-L.; Chuang, S.-T. Molecular Epidemiological and Serological Studies of Bovine Leukemia Virus in Taiwan Dairy Cattle. Front. Vet. Sci. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Buehring, G.C. Natural genetic variations in bovine leukemia virus envelope gene: Possible effects of selection and escape. Virology 2007, 366, 150–165. [Google Scholar] [CrossRef] [Green Version]
- Inoue, E.; Matsumura, K.; Maekawa, K.; Nagatsuka, K.; Nobuta, M.; Hirata, M.; Minagawa, A.; Osawa, Y.; Okazaki, K. Genetic heterogeneity among bovine leukemia viruses in Japan and their relationship to leukemogenicity. Arch. Virol. 2011, 156, 1137–1141. [Google Scholar] [CrossRef]
- Matsumura, K.; Inoue, E.; Osawa, Y.; Okazaki, K. Molecular epidemiology of bovine leukemia virus associated with enzootic bovine leukosis in Japan. Virus Res. 2011, 155, 343–348. [Google Scholar] [CrossRef]
- Notsu, K.; Wiratsudakul, A.; Mitoma, S.; Daous, H.E.; Kaneko, C.; El-Khaiat, H.M.; Norimine, J.; Sekiguchi, S. Quantitative Risk Assessment for the Introduction of Bovine Leukemia Virus-Infected Cattle Using a Cattle Movement Network Analysis. Pathogens 2020, 9, 903. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Escalante, J.C.; Comas-García, A.; Bernal-Silva, S.; Robles-Espinoza, C.D.; Gómez-Leal, G.; Noyola, D.E. Respiratory syncytial virus A genotype classification based on systematic intergenotypic and intragenotypic sequence analysis. Sci. Rep. 2019, 9, 20097. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Rola-Łuszczak, M.; Kubiś, P.; Balov, S.; Moskalik, R.; Choudhury, B.; Kuźmak, J. Molecular characterization of bovine leukemia virus from Moldovan dairy cattle. Arch. Virol. 2017, 162, 1563–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmal, M.; Hellmann, I.; Liu, W.; Keele, B.F.; Perelson, A.S.; Bhattacharya, T.; Gnanakaran, S.; Daniels, M.; Haynes, B.F.; Korber, B.T.; et al. A Signature in HIV-1 Envelope Leader Peptide Associated with Transition from Acute to Chronic Infection Impacts Envelope Processing and Infectivity. PLoS ONE 2011, 6, e23673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamoun, R.Z.; Morisson, M.; Rebeyrotte, N.; Busetta, B.; Couez, D.; Kettmann, R.; Hospital, M.; Guillemain, B. Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J. Virol. 1990, 64, 4180–4188. [Google Scholar] [CrossRef] [Green Version]
- Portetelle, D.; Dandoy, C.; Burny, A.; Zavada, J.; Siakkou, H.; Gras-Masse, H.; Drobecq, H.; Tartar, A. Synthetic peptides approach to identification of epitopes on bovine leukemia virus envelope glycoprotein gp51. Virology 1989, 169, 34–41. [Google Scholar] [CrossRef]
- Licursi, M.; Inoshima, Y.; Wu, D.; Yokoyama, T.; González, E.T.; Sentsui, H. Provirus variants of bovine leukemia virus in naturally infected cattle from Argentina and Japan. Vet. Microbiol. 2003, 96, 17–23. [Google Scholar] [CrossRef]
- Rola-Łuszczak, M.; Grabowska, A.; Szewczyk, B.; Kuźmak, J. Baculovirus expression and potential diagnostic application of the gp51 envelope glycoprotein of genetic mutants of the bovine leukaemia virus. J. Vet. Res. 2019, 63, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Meade, K.G.; Gormley, E.; Doyle, M.B.; Fitzsimons, T.; O’ Farrelly, C.; Costello, E.; Keane, J.; Zhao, Y.; MacHugh, D.E. Innate gene repression associated with Mycobacterium bovis infection in cattle: Toward a gene signature of disease. BMC Genom. 2007, 8, 400. [Google Scholar] [CrossRef] [Green Version]
- Pluta, A.; Albritton, L.M.; Rola-Łuszczak, M.; Kuźmak, J. Computational analysis of envelope glycoproteins from diverse geographical isolates of bovine leukemia virus identifies highly conserved peptide motifs. Retrovirology 2018, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Willems, L.; Thienpont, E.; Kerkhofs, P.; Burny, A.; Mammerickx, M.; Kettmann, R. Bovine leukemia virus, an animal model for the study of intrastrain variability. J. Virol. 1993, 67, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Pomier, C.; Alcaraz, M.; Debacq, C.; Lançon, A.; Kerkhofs, P.; Willems, L.; Wattel, E.; Mortreux, F. Early and transient reverse transcription during primary deltaretroviral infection of sheep. Retrovirology 2008, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, J.P.; Pluta, A.; Rola-Łuszczak, M.; McGowan, S.L.; Finnegan, C.; Heenemann, K.; Carignano, H.A.; Alvarez, I.; Murakami, K.; Willems, L.; et al. Interlaboratory Comparison of Six Real-Time PCR Assays for Detection of Bovine Leukemia Virus Proviral DNA. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [Green Version]
- OIE (World Organization for Animal Health), Chapter 3.4.9. Enzootic Bovine Leukosis in: Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; World Organization for Animal Health (OIE): Paris, France; ISBN 978-92-95108-18-9.
- Rulka, J.; Kubis, P.; Deren, W.B.E. Evaluation of the nested-PCR method for the diagnosis of bovine leukaemia virus (BLV) infection. Bull. Vet. Inst. Pulawy 2001, 45, 11–19. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. 1999, Ser.41, 95–98. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B.M. BBMap: A Fast, Accurate, Splice-Aware Aligner. Available online: https://www.osti.gov/servlets/purl/1241166 (accessed on 10 June 2021).
- Obenchain, V.; Lawrence, M.; Carey, V.; Gogarten, S.; Shannon, P.; Morgan, M. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 2014, 30, 2076–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Origin of Sample | PCR Results 1 | |
|---|---|---|---|
| 444 bp | 993 bp | ||
| J146 blood clot | Khyber Pakhtunkwa (farm H) | - | - |
| J146 mesentery lymph node | - | - | |
| J760 blood coat (6Pak) | + | - | |
| J116 blood clot | - | - | |
| J116 scapular lymph node | - | - | |
| J116 lung lymph node (2Pak) | + | - | |
| J116 liver | - | - | |
| J116 spleen | - | - | |
| J116 brain tissue | - | - | |
| P2 | + | + | |
| P4 | + | - | |
| P5 | + | + | |
| P6 | + | + | |
| P7 | + | - | |
| P29 | + | + | |
| P30 | + | + | |
| P474 | + | + | |
| P475 | + | + | |
| P479 | + | + | |
| P488 | + | + | |
| P492 | + | + | |
| P496 | + | + | |
| P500 | + | + | |
| P504 | + | + | |
| P506 | + | + | |
| G6 | Gilgit Baltisan (farm G1) | - | - |
| G23 | Gilgit Baltisan (farm G2) | + | - |
| L348 | Punjab (farm B) | + | + |
| L351 | + | - | |
| L364 | + | + | |
| L367 | + | + | |
| L368 | + | + | |
| L371 | + | + | |
| L376 | + | + | |
| L381 | + | + | |
| L382 | + | + | |
| L384 | + | - | |
| L387 | + | - | |
| L391Q | + | + | |
| L392 | + | + | |
| Leader Peptide | G Epitope | Zn Binding Peptide | A | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ND2 | D D’ Epitope | Epitope | ||||||||||
| 4 | 12 | 28 | 29 | 48 | 74 | 82 | 144 | 254 | 264 | 267 | 291 | |
| FLKBLV | K | Q | C | R | A | K | S | I | S | P | R | A |
| P5 | E | . | S | . | . | . | . | . | . | . | . | . |
| P6 | E | . | S | . | . | . | . | . | . | . | . | . |
| P479 | E | . | S | . | . | . | . | . | . | . | . | . |
| P488 | E | . | S | . | . | . | . | . | . | . | . | . |
| P492 | E | . | S | . | . | . | . | . | . | . | . | . |
| P475 | E | . | S | . | . | . | . | . | . | . | . | . |
| P2 | E | . | S | . | . | . | . | . | . | . | . | . |
| P29 | E | . | S | . | . | . | . | . | . | . | . | . |
| P30 | E | . | S | . | . | . | . | . | . | . | . | . |
| P504 | E | . | S | . | . | . | . | . | . | . | . | T |
| P474 | E | . | . | . | . | . | . | . | . | . | . | . |
| P496 | E | . | . | . | . | . | . | . | . | . | . | . |
| P500 | E | . | . | . | . | . | . | . | . | . | . | . |
| P506 | E | . | . | . | . | . | . | . | . | . | . | . |
| G23 | ~ | ~ | ~ | ~ | ~ | ~ | ~ | . | ~ | ~ | ~ | ~ |
| 2Pak | ~ | ~ | ~ | ~ | ~ | ~ | ~ | . | ~ | ~ | ~ | ~ |
| 6Pak | ~ | ~ | ~ | ~ | ~ | ~ | ~ | . | ~ | ~ | ~ | ~ |
| L348 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L371 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L364 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L367 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L382 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L392 | E | E | . | Q | T | R | F | T | L | S | K | . |
| L376 | E | E | . | Q | T | R | F | T | L | . | K | . |
| L368 | E | E | . | Q | T | R | F | T | L | . | K | . |
| L381 | E | E | . | Q | T | R | F | T | L | . | K | . |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rola-Łuszczak, M.; Sakhawat, A.; Pluta, A.; Ryło, A.; Bomba, A.; Bibi, N.; Kuźmak, J. Molecular Characterization of the env Gene of Bovine Leukemia Virus in Cattle from Pakistan with NGS-Based Evidence of Virus Heterogeneity. Pathogens 2021, 10, 910. https://doi.org/10.3390/pathogens10070910
Rola-Łuszczak M, Sakhawat A, Pluta A, Ryło A, Bomba A, Bibi N, Kuźmak J. Molecular Characterization of the env Gene of Bovine Leukemia Virus in Cattle from Pakistan with NGS-Based Evidence of Virus Heterogeneity. Pathogens. 2021; 10(7):910. https://doi.org/10.3390/pathogens10070910
Chicago/Turabian StyleRola-Łuszczak, Marzena, Ali Sakhawat, Aneta Pluta, Anna Ryło, Arkadiusz Bomba, Nazia Bibi, and Jacek Kuźmak. 2021. "Molecular Characterization of the env Gene of Bovine Leukemia Virus in Cattle from Pakistan with NGS-Based Evidence of Virus Heterogeneity" Pathogens 10, no. 7: 910. https://doi.org/10.3390/pathogens10070910
APA StyleRola-Łuszczak, M., Sakhawat, A., Pluta, A., Ryło, A., Bomba, A., Bibi, N., & Kuźmak, J. (2021). Molecular Characterization of the env Gene of Bovine Leukemia Virus in Cattle from Pakistan with NGS-Based Evidence of Virus Heterogeneity. Pathogens, 10(7), 910. https://doi.org/10.3390/pathogens10070910