1. Introduction
The Gram-negative bacterium
Helicobacter pylori (
H. pylori) infects the gastric mucosa and is associated with different human diseases, e.g., chronic active gastritis, peptic ulcer disease, gastric cancer or mucosa-associated lymphoid tissue (MALT)-lymphoma [
1]. In Germany,
H. pylori infections are commonly treated first-line by a triple therapy consisting of two different antibiotics, usually amoxicillin and clarithromycin or metronidazole, and a proton pump inhibitor (PPI) [
2]. However, this standard first-line therapy for the eradication of
H. pylori faces a serious problem due to the increasing rates of resistance against these antibiotics, which may result in therapy failure. Thus, a quadruple therapy consisting of bismuth in combination with amoxicillin, a PPI and a quinolone, e.g., levofloxacin, is now often recommended [
3,
4].
Quinolones target the DNA gyrase, which is an essential enzyme in bacterial replication that can create negative and positive supercoils in DNA by transiently introducing double-strand breaks in an ATP-dependent reaction [
5]. DNA gyrase is heterotetrameric consisting of two A and two B subunits, respectively. The A subunit contains a tyrosine residue in the active center that covalently binds the newly generated 5′ termini of DNA during the cleavage reaction, while the B subunit harbors the ATPase domain responsible for DNA cleavage and ligation. Quinolones were first reported to target the A subunit of the DNA gyrase in
H. pylori [
6]. However, it has been also suggested that mutations in the B subunit may contribute to quinolone resistance [
7,
8,
9]. In particular, resistance against quinolones can be achieved by single point mutations in the so-called quinolone resistance-determining region (QRDR) of the
gyrA gene of
H. pylori [
10,
11,
12]. The mutations conferring resistance result in specific amino acid exchanges at codons 87 and/or 91 in QRDR, which, in turn, lead to a weaker binding of the antibiotic. The most common amino acid exchanges appear to be N87K and D91G, D91N and D91Y (
Table 1) [
13,
14,
15]. The N87K mutation alone confers high resistance to levofloxacin and gatifloxacin [
7]. Depending on the geographical region, in which the isolates were obtained, additional but rarely occurring amino acid exchanges have been detected, i.e., N87I, N87R, D91M, D91C, D91A, and N87H in combination with D91M [
16,
17]. However, the contribution of these mutations to antibiotic resistance has not been tested in all cases.
A common method to examine resistance to quinolone is via hybridization probes on stripes. This procedure is used in the commercial kit GenoType HelicoDR from Hain Lifesciences (Nehren, Germany). Despite the advantageous suitability of this kit for cell culture testing, it requires, in addition to a thermal cycler, an incubator and an apparatus for the visual evaluation of the colour reactions on the stripes. Moreover, this test takes more than five hours to complete. Another resistance-determining technique is the whole-genome sequencing of
H. pylori via the Next Generation Sequencing (NGS) methodology. NGS is especially useful in the identification of new resistance mutations. However, this approach is very expensive and time-consuming for routine examinations [
18]. Above all, for the treatment of
H. pylori infections, it is important to be able to distinguish between wild-type (WT) and mutant (MT) QRDR sequences. These requirements can be met by an RT-PCR-based assay, which can be performed in only a few hours. Furthermore, compared to any dot blot hybridization method, the sensitivity obtained using real-time polymerase chain reaction (RT-PCR) is much higher, because DNA amplification can detect very low copy numbers of the specific DNA sequence.
In this work, we developed an assay based on melting curve analyses using RT-PCR to distinguish between the known WT and the most prevalent MT QRDR sequences (
Table 1) [
13,
14,
15]. In contrast to previous approaches, by cultivating the bacteria to determine the resistance pattern (E-test), which usually takes about five days, genotyping with RT-PCR can provide consistent results within one day, thus saving time, money and additional biopsy procedures for culture. This method allows for the reliable identification of
H. pylori strains in patients, which are resistant to quinolones using an RT-PCR assay based on Förster resonance energy transfer (FRET), and the determination of different melting point temperatures (T
m) of fluorescently labeled probes that are complementary to the relevant QRDR sequence. The assay only requires a set of three different probes, one WT sequence and two mutant sequences to reliably determine if resistant
H. pylori is present or not. Our method provides a rapid and cost-effective new tool for the improved detection of all prevalent resistance-associated mutations against quinolones in the QRDR and, in addition, can discriminate between MT and WT sequences. Thus, the optimal combination of antibiotics for the patient can be determined before treatment.
2. Results and Discussion
The aim of this study was to develop a fast and low-cost assay, which can unambiguously distinguish between
H. pylori strains from patient biopsies that are sensitive or resistant to quinolone antibiotics. Using this strategy, inefficient treatment of patients can be avoided with savings in time, money and additional biopsy procedures for culture. A previously published assay detected
gyrA mutations in
H. pylori by allele-specific PCR [
19]. However, this method requires the application of a set of eight different allele-specific primers. After PCR amplification, the PCR products have to be visually analyzed by agarose gel electrophoresis and are distinguished according to their size.
A RT-PCR assay, developed by Glocker and Kist [
14], aimed to detect WT and MT QRDR sequences bya Förster resonance energy transfer (FRET)-based RT-PCR assay, using a labeled anchor probe in combination with a labeled mutation probe for each codon. The assay established here combines that approach with a method used for the detection of clarithromycin resistance in
H. pylori, as reported by Schabereiter-Gurtner et al. [
20]. Only two fluorescent probes, specific to the mutant genotypes corresponding to MT1 and MT3, respectively (
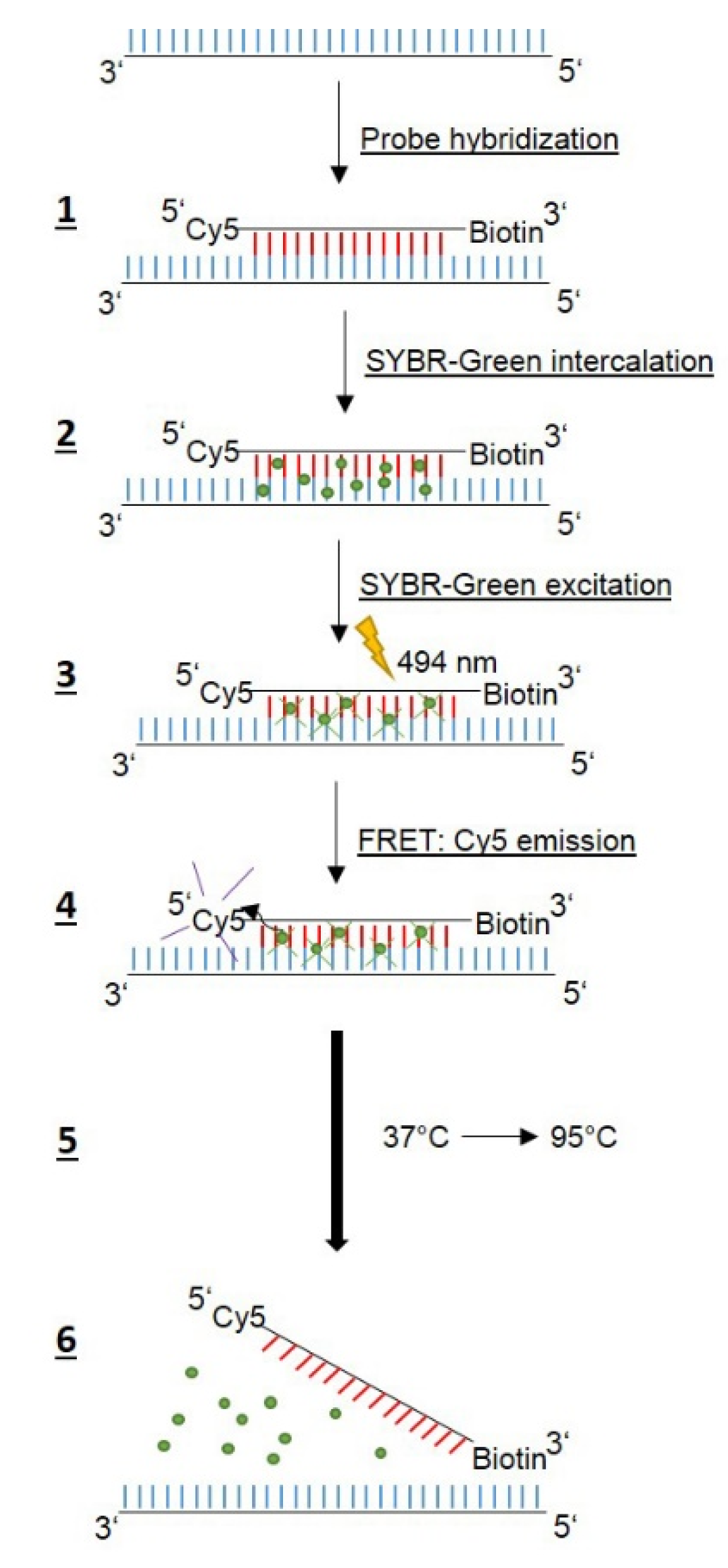
Table 1), are needed, but no anchor probes, since here FRET is based on the intercalation and excitation of SYBR-Green in the amplified DNA, followed by emission via the Cyanine5 (Cy5) label of the probe (
Figure 1).
To find reliable assay conditions, we first generated plasmid DNAs harboring two defined WT sequences (WT1 and WT2) and six MT QRDR sequences (MT1 to MT6) as shown in
Table 1 and
Table 2, respectively. We decided not to use different fluorescent labels, which would allow two probes to be used at the same time to avoid misinterpretations, since all probes bind to the same region. A single-stranded DNA probe complementary to the QRDR WT1 sequence, labeled with Cy5 at the 5′ end and biotin at the 3′ end to prevent polymerization, was used for melting curve analyses (
Table 3). SYBR-green, a dye intercalating into double-stranded DNA was added to the reaction mix. RT-PCR and subsequent melting curve analysis (
Table 4) was performed according to the scheme shown in
Figure 1. The Cy5 WT1 probe binds specifically to the region harboring codons 87 and 91 of the amplified DNA, while SYBR-green intercalates into the double-stranded DNA. The excitation of SYBR-green at 494 nm results in FRET to the Cy5 label, and Cy5 emission can be detected at 670 nm. Applying a temperature-gradient (37 °C to 95 °C) leads to dissociation of the Cy5 probe at a specific temperature and, thus, the FRET signal abates. Accordingly, mutations in the QRDR template DNA will give rise to lower T
m values due to the weaker binding of the Cy5 probe, which is 100% complementary to only the WT1 QRDR.
Using the Cy5 WT1 probe (
Table 3), corresponding to the WT1 QRDR sequence, resulted in a high background noise, which made analysis of the melting curves difficult, and no signal could be detected for MT3 (data not shown). Thus, we designed new Cy5 probes corresponding to MT1 and MT3 (
Table 3) and performed RT-PCR reactions to determine the T
m-values. With both probes, the two WT sequences, as well as all MT QRDR sequences (
Table 1), could be detected and exhibited different T
m values (
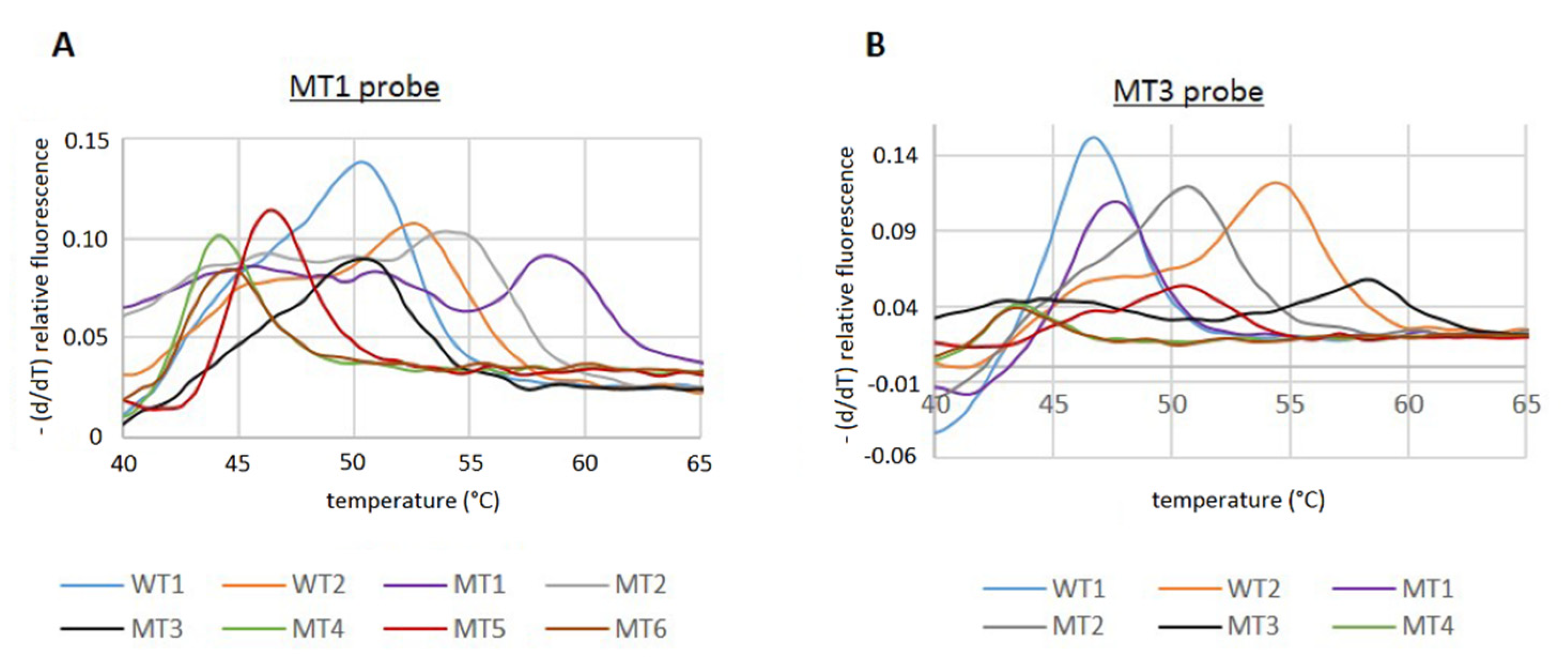
Figure 2).
However, since the T
m values of some of the MT PCR products were very similar compared to the WT, it was not possible to unambiguously distinguish between all MT and WT sequences with just one of the Cy5 probes. Using the MT3 probe, no discrimination of the T
m of the WT1/MT1, MT2/MT5, and MT4/MT6 was possible. However, the MT1 probe allowed us to distinguish between the WT1 and MT1 sequence, but the discrimination of WT2/MT2, WT1/MT3, and MT4/MT6 could not be achieved (
Figure 2;
Table 5).
Nevertheless, our setup allowed for a general discrimination between WT and MT sequences. Since only patients infected with
H. pylori WT strains, which are susceptible to quinolones, can be successfully treated with the antibiotic, it is sufficient to be able to separate the WT sequences from all MT sequences. Thus, by using our two MT Cy5 probes in parallel, it is possible to assign a sequence either to a WT or MT phenotype, respectively. If a patient sample is tested with the MT1 probe and a T
m of ca. 50 °C is determined, it will not be clear whether the
H. pylori strain harbors the WT1 or MT3 QRDR sequence (
Table 5). Thus, a second RT-PCR reaction with the MT3 probe can be performed for identification. However, with the MT3 probe, the WT1 will have a T
m of 46.86 °C, the T
m of the MT3 genotype will be increased to 58.2 °C due to the 100% complementarity of the MT3 probe. The same procedure can be applied if T
m = 52.7 °C, and no distinction between WT2 and MT2 can be made, since the T
m values of the two sequences differ by 4 °C when the MT3 probe is used. On the other hand, no distinction between WT1 and MT1 can be made if only the MT3 probe is used. However, by applying the MT1 probe, the T
m values are easily distinguishable (49.99 °C vs. 58.49 °C).
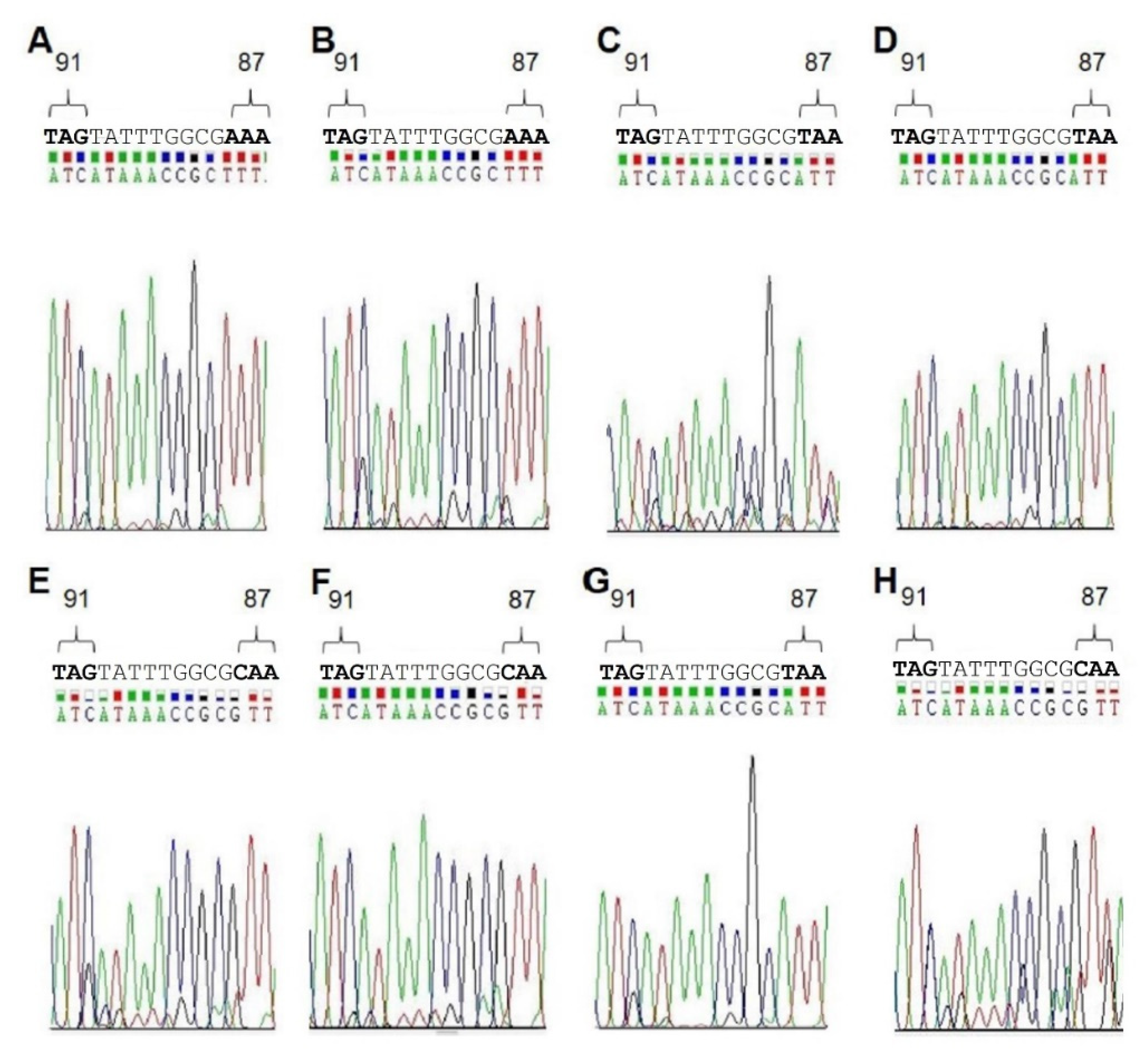
Finally, to further examine the validity of our results, we analyzed DNA isolated from biopsies taken from patients with known
H. pylori infections. Each sample was tested with the MT1, as well as with the MT3 probe. In addition, to confirm the results obtained by melting curve analyses, the patient DNAs were sequenced to determine the QRDR genotype (
Table 6,
Figure 3). All sequencing data were in agreement with the genotype determined by RT-PCR (
Table 6,
Figure 3). As negative controls, and to determine the specificity of our test, we isolated biopsy DNA from six patients known not to be infected with
H. pylori and performed RT-PCR with the QRDR primers. As expected, no QRDR sequence could be detected in all cases (
Table 6, bottom,
Supplementary Data, Figure S1A,B), which confirms the good specificity of this test.
3. Material and Methods
Plasmids. The
gyrA QRDR sequence of two antibiotics-susceptible clinical WT strains were utilized, WT1 (GenBank accession no. CP026515.1) and WT2 (GenBank accession no. CP026515.1). The two WT sequences differ only in the codon at position 87 (
Table 1, top). The WT1
gyrA sequence was amplified by PCR with the FP and RP GyrA WT1 primers (
Table 2) using sequenced patient DNA as a template, and cloned into the vector pUC57 via the restriction sites XbaI and BamHI (Genscript, Piscataway, NJ, USA). WT2 and six QRDR mutations (named MT1 to MT6) were introduced by site-directed mutagenesis in the WT1 sequence (
Table 1). The primers for mutagenesis shown in
Table 2 were applied according to the Quick Change mutagenesis protocol (Agilent Technologies, Santa Clara, CA, USA) and confirmed by standard sequencing (GATC Biotech AG, Konstanz, Germany). Plasmids were propagated in
Escherichia coli strains XL1-Blue or Top10 and isolated using the QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany).
Patient DNA isolation, amplification and sequencing. DNA of patients infected with
H. pylori was isolated from formalin fixed, paraffin embedded (FFPE) gastric biopsies (ethics statement number: 322_21 Bc, University of Erlangen, Germany) using the Maxwell
® 16 LEV Blood Kit (Promega GmbH, Mannheim, Germany). Usually, six thin sections of 2 µm thickness were used for DNA extractions. The concentration was measured by UV-Vis spectroscopy at 260 nm. Approximately 200–400 ng of isolated DNA was used to amplify the QRDR sequence by PCR using the primers gyrA-FP and gyrA-RP (
Table 3) with the PCR program depicted in
Table 4 without the melting curve step. To avoid misinterpretation of the results, lower DNA concentrations are not recommended. The amplified QRDR fragments were sequenced using gyrA-RP as a sequencing primer (GATC Biotech AG, Konstanz, Germany).
Melting curve analyses. All plasmid and patient DNAs were analyzed by melting curve analyses. Primers and probes (metabion GmbH, Planegg/Steinkirchen, Germany) used are shown in
Table 3. The reaction mix contained 0.625 µM forward primer (FP), 0.19 µM reverse primer (RP), 0.5 µM probe, 3 mM MgCl
2, 2 µL LightCycler FastStart DNA Master SYBR green I (Roche Diagnostics, Mannheim, Germany) and 40 ng of template DNA in 20 µL reactions. Samples were analyzed using a Cobas Z 480 Analyzer (Roche Diagnostics, Mannheim, Germany) according to the RT PCR program shown in
Table 4. The results were evaluated with the
LightCycler® 480 software 1.5.1 (Roche Diagnostics, Mannheim, Germany). Several independent melting curve analyses were performed for each QRDR sequence and the average of the melting temperature (T
m), as well as the standard deviation (S) were estimated according to Equations (1) and (2):
n, number of measurements; T
m, melting temperature;
, average of melting temperature of n measurements; S, standard deviation.



 ,
, 



{kind=link}
{kind=link}
{kind=link}