




Dynamic Analysis of microRNAs from Different Life Stages of Rhipicephalus microplus (Acari: Ixodidae) by High-Throughput Sequencing



Abstract
:1. Introduction
2. Results
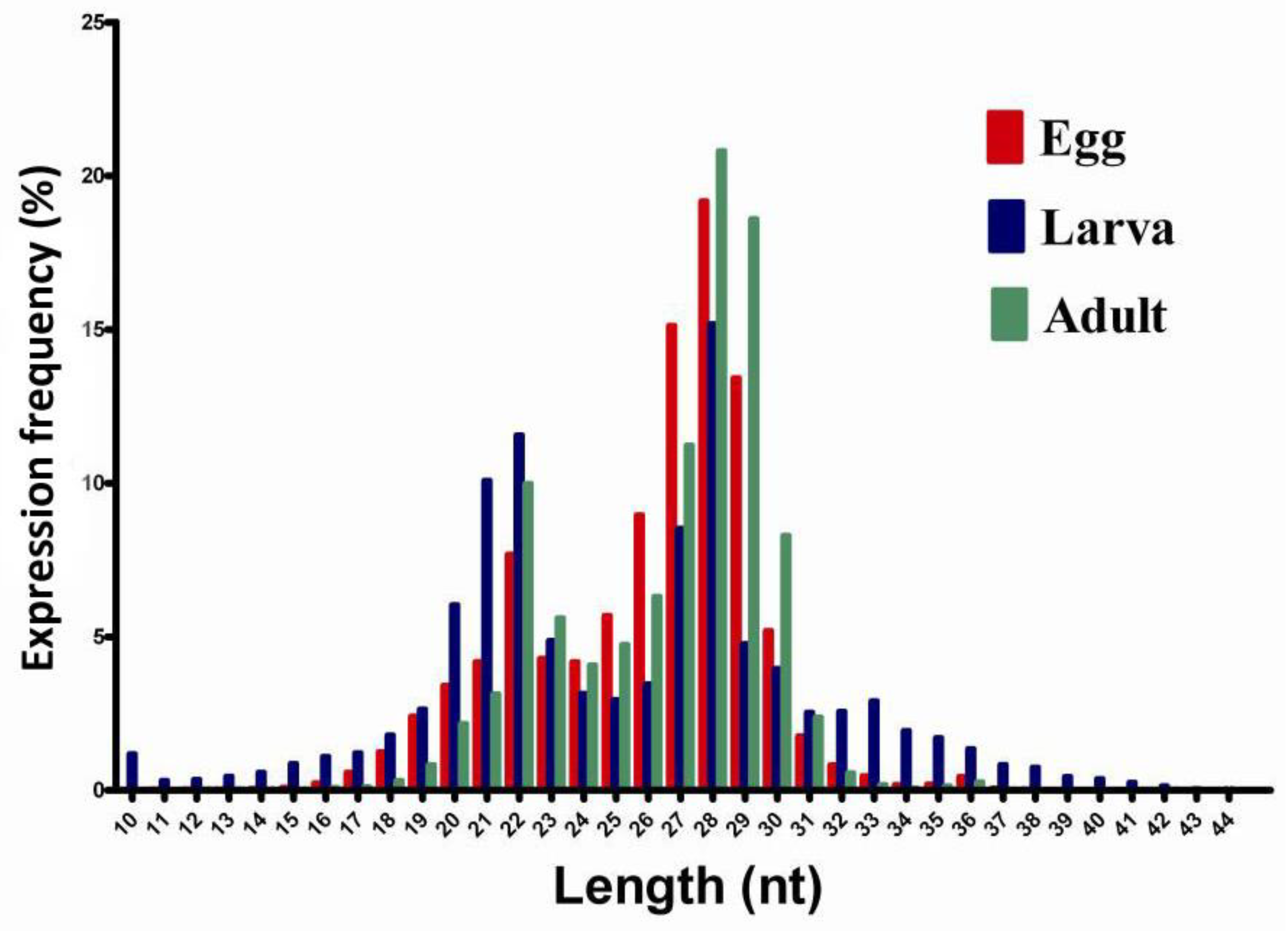
2.1. Small RNA Library Construction and Solexa Sequencing
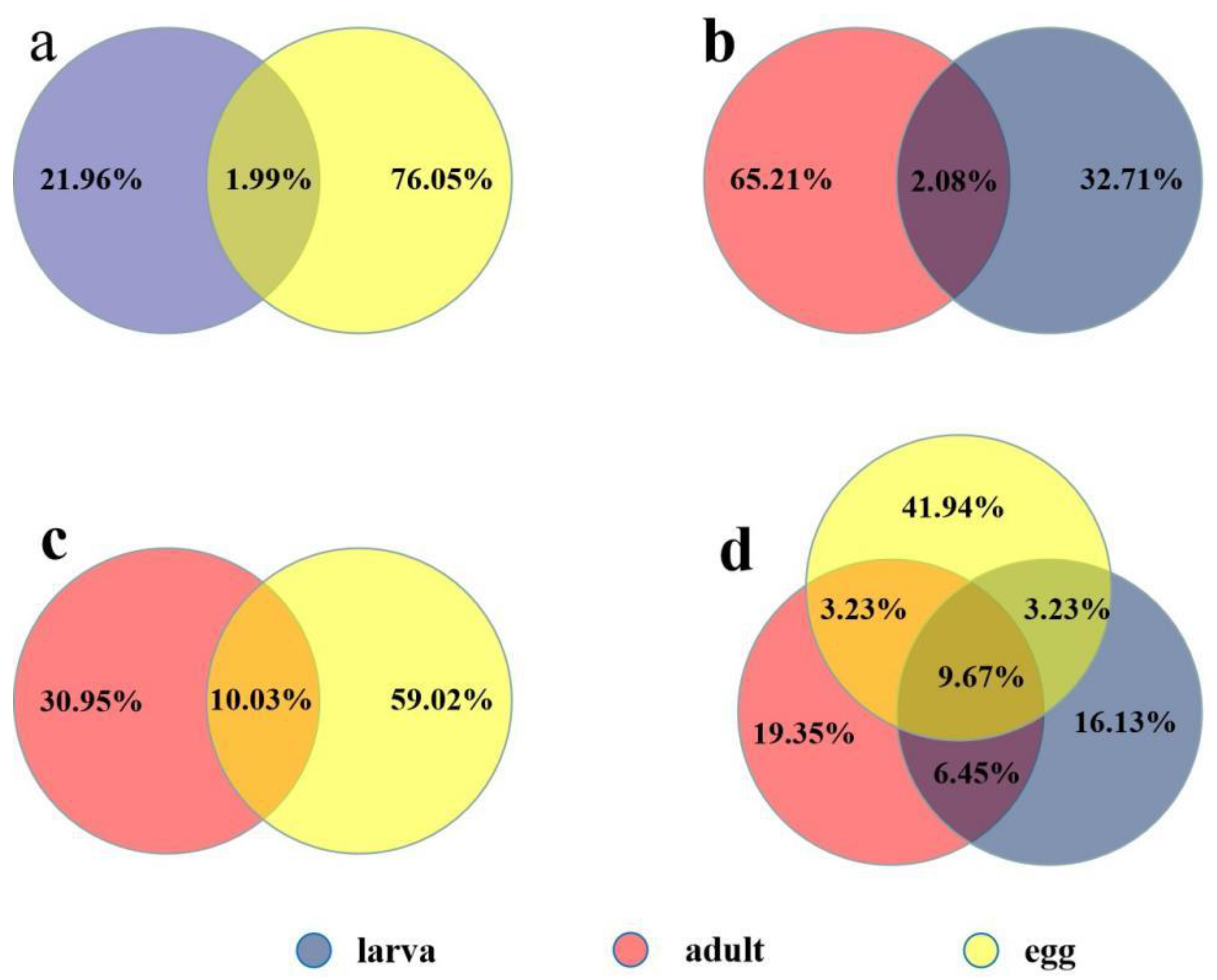
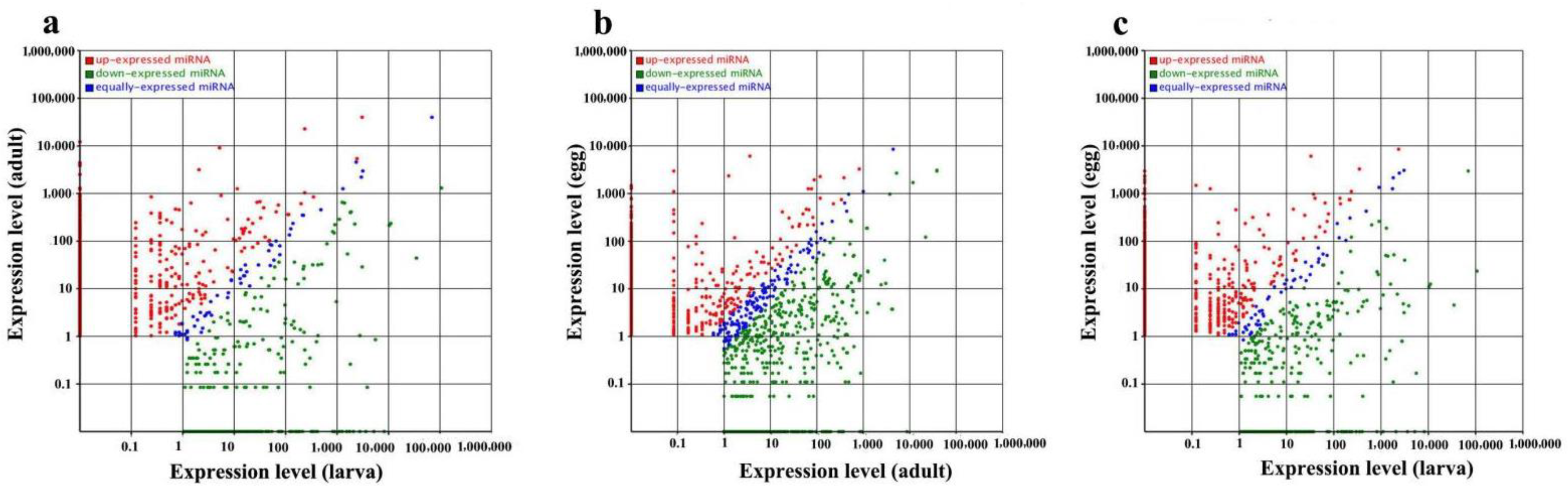
2.2. Known Conserved MicroRNAs and Differential Expression at Different Developmental Stages
2.3. Identification of Novel microRNA Candidates
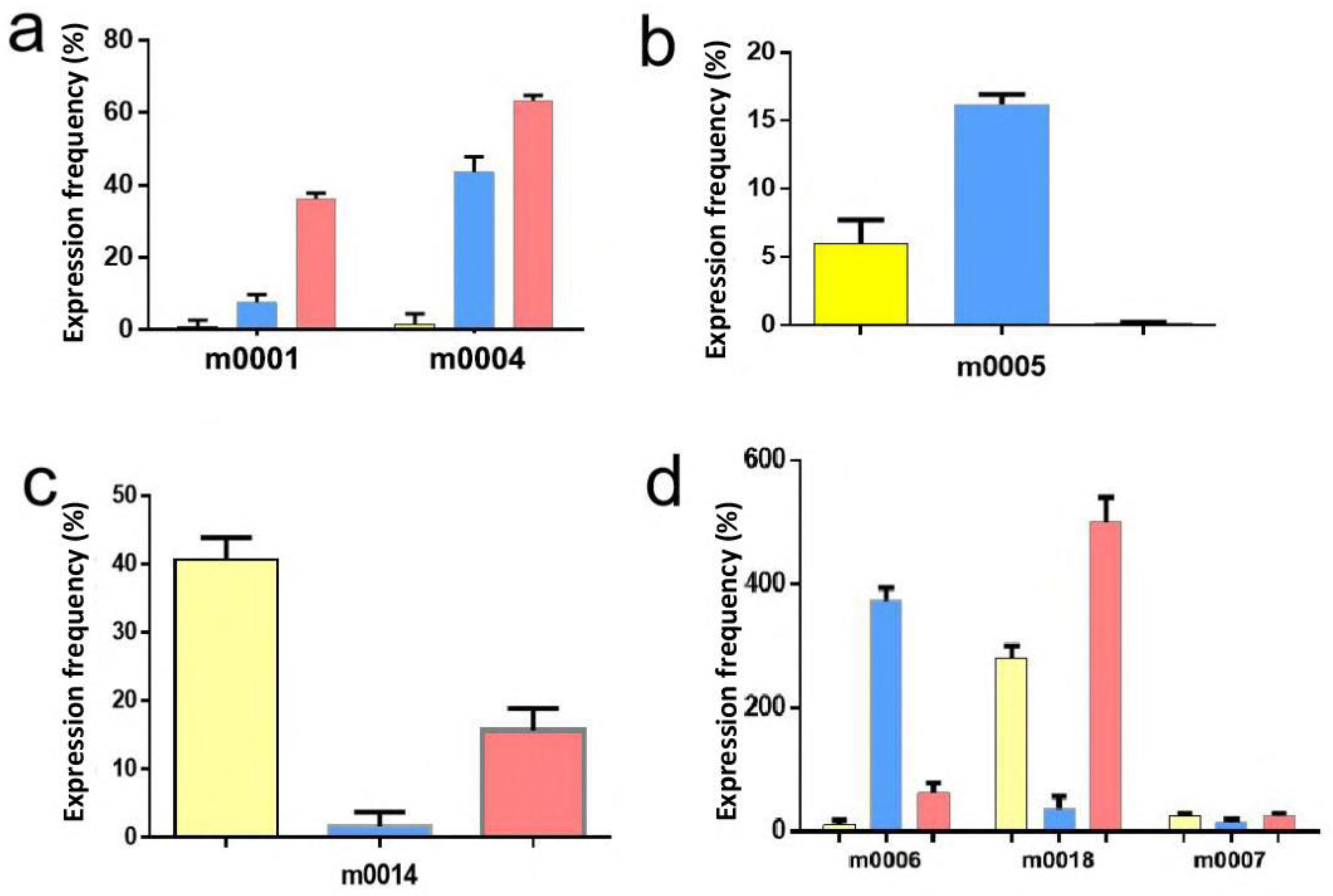
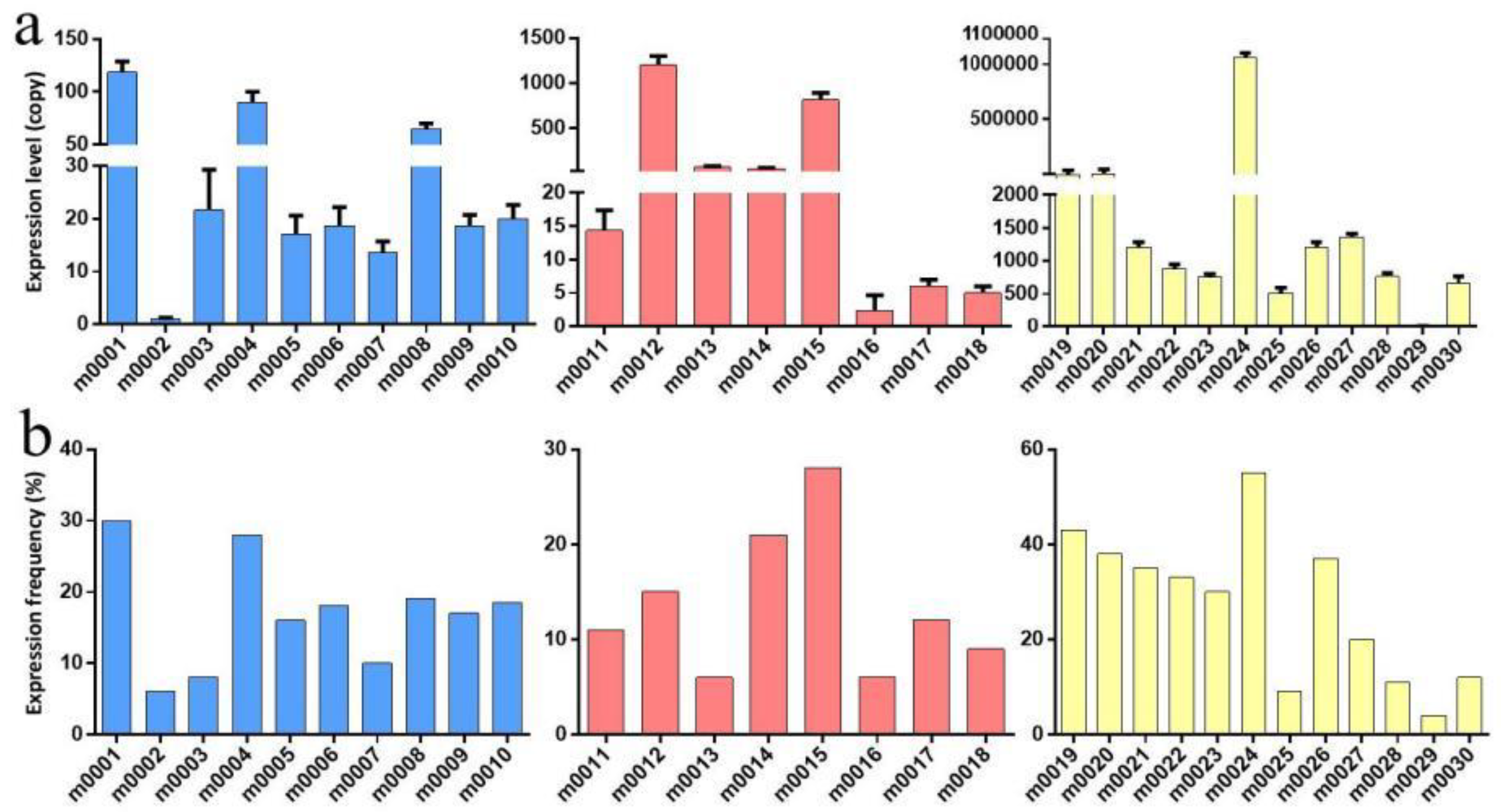
2.4. Validation of MicroRNA Expression by Quantitative RT–PCR
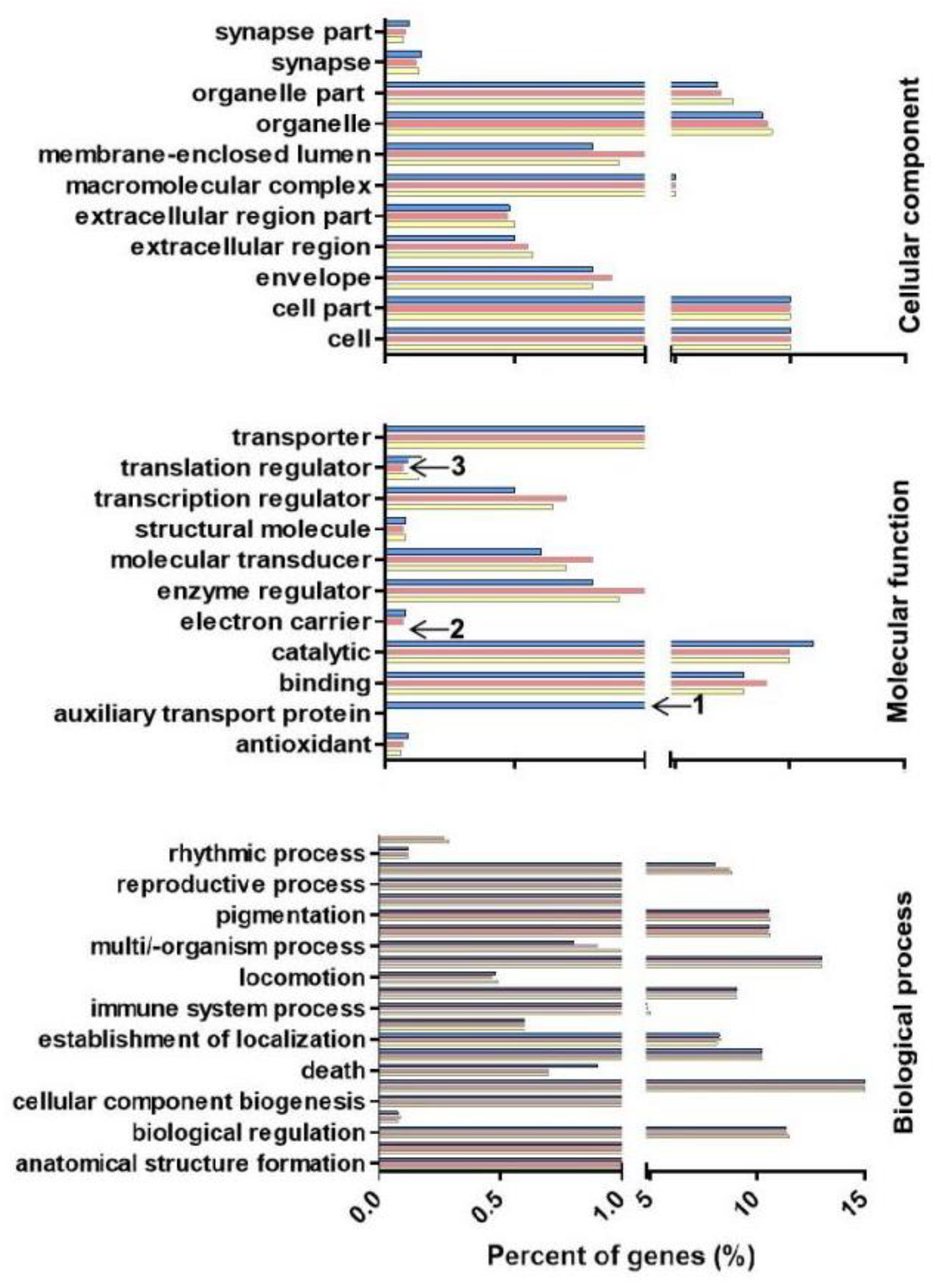
2.5. MicroRNA Target Gene Prediction and GO Enrichment and KEGG Pathway Analyses
3. Discussion
4. Materials and Methods
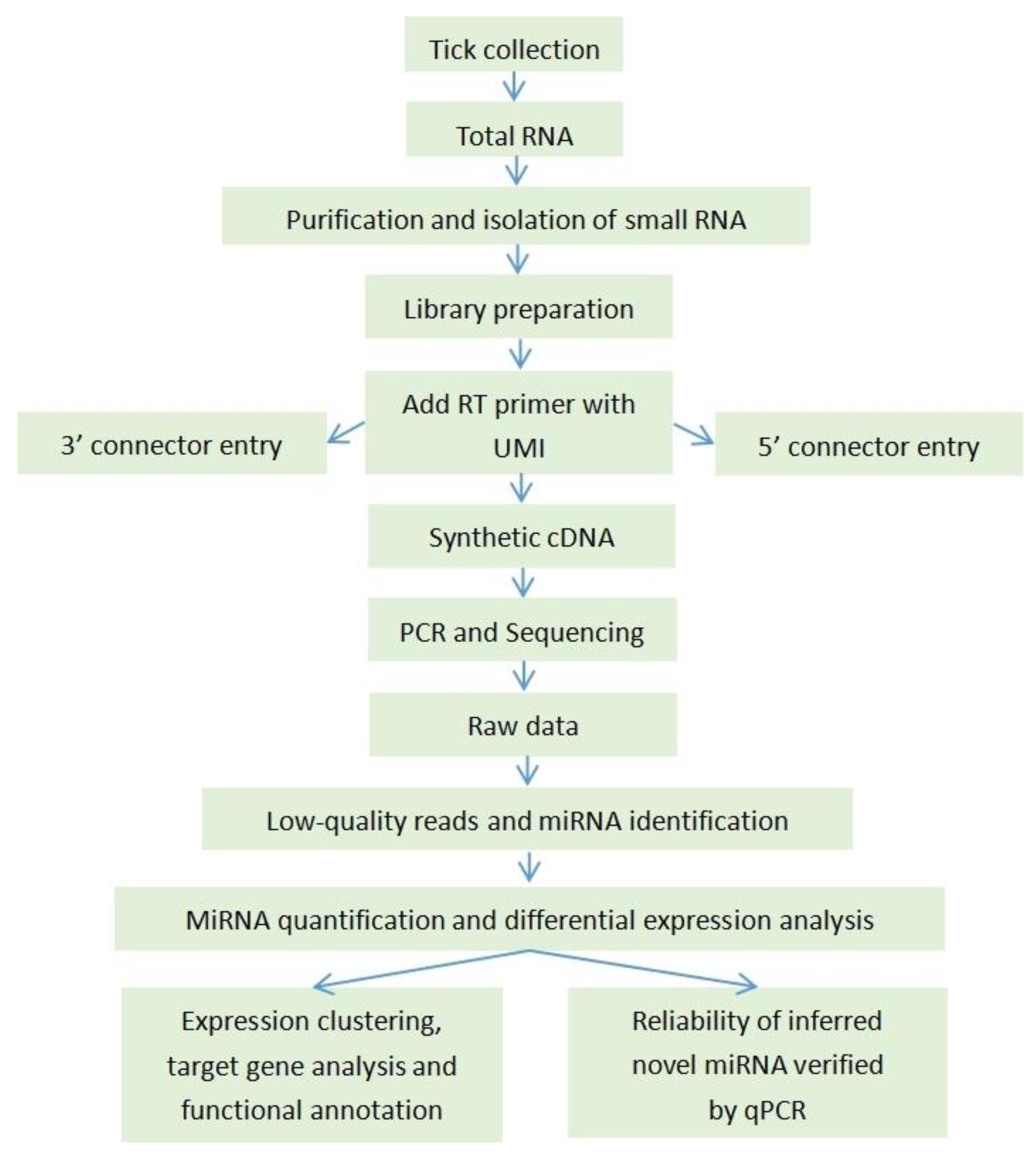
4.1. The Experimental Analysis Workflow
4.2. Tick Collection and RNA Extraction and Library Preparation
4.3. Small RNA Isolation and High-Throughput Sequencing
4.4. Small RNA Bioinformatics Analysis
4.5. Target Prediction for Novel miRNA Candidates
4.6. Differential Expression of Known miRNAs and GO Enrichment and KEGG Pathway Analyses
4.7. Real-Time Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujisaki, K.; Kawazu, S.; Kamio, T. The taxonomy of the bovine Spp. Parasitol. Today 1994, 10, 31–33. [Google Scholar] [CrossRef]
- Luo, J.; Chen, F.; Lu, W.; Guan, G.; Ma, M.; Yin, H. Experimental transmission of an unnamed bovine Babesia by Hyalomma spp., Haemaphysalis longicornis and Boophilus microplus. Vet. Parasitol. 2003, 116, 115–124. [Google Scholar] [CrossRef]
- Jongejan, F.; Uilenberg, G. The global importance of ticks. Parasitology 2004, 129, S3–S14. [Google Scholar] [CrossRef]
- Ahmed, J.S.; Luo, J.; Schnittger, L.; Seitzer, U.; Jongejan, F.; Yin, H. Phylogenetic Position of Small-Ruminant Infecting Piroplasms. Ann. N. Y. Acad. Sci. 2006, 1081, 498–504. [Google Scholar] [CrossRef]
- Yin, H.; Schnittger, L.; Luo, J.; Seitzer, U.; Ahmed, J.S. Ovine theileriosis in China: A new look at an old story. Parasitol. Res. 2007, 101, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.T.; Badra, S.J.; Pereira, L.E.; Szabó, M.P. Report on ticks collected in the Southeast and Mid-West regions of Brazil: Analyzing the potential transmission of tick-borne pathogens to man. Rev. Da Soc. Bras. De Med. Trop. 1999, 32, 613–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Peña, A.; Bouattour, A.; Camicas, J.L.; Guglielmone, A.; Horak, I.; Jongejan, F.; Latif, A.; Pegram, R.; Walker, A.R. The known distribution and ecological preferences of the tick subgenus Boophilus (Acari: Ixodidae) in Africa and Latin America. Exp. Appl. Acarol. 2006, 38, 219–235. [Google Scholar] [CrossRef]
- Luo, J.; Wu, F.; Liu, W.; Ren, Q.; Diao, P.; Guan, G.; Luo, J.; Yin, H.; Liu, G. A Novel MicroRNA and the Target Gene TAB2 Can Regulate the Process of Sucking Blood in and the Spawn Rate of Hyalomma asiaticum (Acari: Ixodidae) Ticks. Front. Immunol. 2022, 13, 930532. [Google Scholar] [CrossRef]
- Farh, K.K.H.; Grimson, A.; Jan, C.; Lewis, B.P.; Johnston, W.K.; Lim, L.P.; Burge, C.B.; Bartel, D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaubert, S.; Mereau, A.; Antoniewski, C.; Tagu, D. MicroRNAs in Drosophila: The magic wand to enter the Chamber of Secrets? Biochimie 2007, 89, 1211–1220. [Google Scholar] [CrossRef]
- Sempere, L.F.; Martinez, P.; Cole, C.; Baguñà, J.; Peterson, K.J. Phylogenetic distribution of microRNAs supports the basal position of acoel flatworms and the polyphyly of Platyhelminthes. Evol. Dev. 2007, 9, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Glazov, E.A.; Cottee, P.A.; Barris, W.C.; Moore, R.J.; Dalrymple, B.P.; Tizard, M.L. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008, 18, 957–964. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Cai, P.; Jiang, N.; Wang, H.; Chen, Q. Identification and characterization of microRNAs and endogenous siRNAs in Schistosoma japonicum. BMC Genom. 2010, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Taft, R.J.; Asgari, S. An insect virus-encoded microRNA regulates viral replication. J. Virol. 2008, 82, 9164–9170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrero, R.A.; Keeble-Gagnère, G.; Zhang, B.; Moolhuijzen, P.; Ikeo, K.; Tateno, Y.; Gojobori, T.; Guerrero, F.D.; Lew-Tabor, A.; Bellgard, M. Evolutionary conserved microRNAs are ubiquitously expressed compared to tick-specific miRNAs in the cattle tick Rhipicephalus (Boophilus) microplus. BMC Genom. 2011, 12, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zou, Q.; Song, F.; Wang, X.; Shen, X.J. The regulation of silkworm fibroin L chain production by miRNA-965 and miRNA-1926 in insect cells. Russ. J. Bioorg. Chem. 2012, 38, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Ren, Q.; Liu, W.; Qiu, X.; Zhang, G.; Tan, Y.; Cao, R.; Yin, H.; Luo, J.; Li, X.; et al. MicroRNA-1 Expression and Function in Hyalomma Anatolicum Anatolicum (Acari: Ixodidae) Ticks. Front. Physiol. 2021, 12, 596289. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Liu, F.; Vongsangnak, W.; Jing, Q.; Shen, B. Performance comparison and evaluation of software tools for microRNA deep-sequencing data analysis. Nucleic Acids Res. 2012, 40, 4298–4305. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540. [Google Scholar] [CrossRef] [Green Version]
- Wienholds, E.; Plasterk, R.H. MicroRNA function in animal development. FEBS Lett. 2005, 579, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Zamore, P.D. Beginning to understand microRNA function. Cell Res. 2007, 17, 661–663. [Google Scholar] [CrossRef]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef]
- Dong, Q.H.; Han, J.; Yu, H.P.; Wang, C.; Zhao, M.Z.; Liu, H.; Ge, A.J.; Fang, J.G. Computational Identification of MicroRNAs in Strawberry Expressed Sequence Tags and Validation of Their Precise Sequences by miR-RACE. J. Hered. 2012, 103, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.Z.; Xia, H.; Frazier, T.P.; Yao, Y.Y.; Bi, Y.P.; Li, A.Q.; Li, M.J.; Li, C.S.; Zhang, B.H.; Wang, X.J. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant. Biol. 2010, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, D.; Li, Q.; Zhao, P.; Xiang, Z.; Xia, Q. MicroRNAs of Bombyx mori identified by Solexa sequencing. BMC Genom. 2010, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Cui, H.; Jiang, S.; Huang, Y.; Huang, X.; Wei, S.; Xu, W.; Qin, Q. Identification of a novel marine fish virus, Singapore grouper iridovirus-encoded microRNAs expressed in grouper cells by Solexa sequencing. PLoS ONE 2011, 6, e19148. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Deng, B.; Qiao, M.; Zheng, R.; Chai, J.; Ding, Y.; Peng, J.; Jiang, S. Solexa sequencing identification of conserved and novel microRNAs in backfat of Large White and Chinese Meishan pigs. PLoS ONE 2012, 7, e31426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathjen, T.; Pais, H.; Sweetman, D.; Moulton, V.; Munsterberg, A.; Dalmay, T. High throughput sequencing of microRNAs in chicken somites. FEBS Lett. 2009, 583, 1422–1426. [Google Scholar] [CrossRef]
- Li, T.; Wu, R.; Zhang, Y.; Zhu, D. A systematic analysis of the skeletal muscle miRNA transcriptome of chicken varieties with divergent skeletal muscle growth identifies novel miRNAs and differentially expressed miRNAs. BMC Genom. 2011, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.Z. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Townley-Tilson, W.H.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar] [CrossRef] [Green Version]
- Parrish, J.Z.; Xu, P.; Kim, C.C.; Jan, L.Y.; Jan, Y.N. The microRNA bantam Functions in Epithelial Cells to Regulate Scaling Growth of Dendrite Arbors in Drosophila Sensory Neurons. Neuron 2009, 63, 788–802. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.W.; Slattery, M.; Mann, R.S. Transcription factor choice in the Hippo signaling pathway: Homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 2009, 23, 2307–2319. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J.; Cohen, S.M. The Hippo Pathway Regulates the bantam microRNA to Control Cell Proliferation and Apoptosis in Drosophila. Cell 2006, 126, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Kadener, S.; Menet, J.S.; Sugino, K.; Horwich, M.D.; Weissbein, U.; Nawathean, P.; Vagin, V.V.; Zamore, P.D.; Nelson, S.B.; Rosbash, M. A role for microRNAs in the Drosophila circadian clock. Genes Dev. 2009, 23, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Wang, Q.; Cobb, G.P.; Anderson, T.A. Computational identification of microRNAs and their targets. Comput. Biol. Chem. 2006, 30, 395–407. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. CMLS 2006, 63, 246–254. [Google Scholar] [CrossRef]
- Ji, Z.; Wang, G.; Xie, Z.; Wang, J.; Zhang, C.; Dong, F.; Chen, C. Identification of Novel and Differentially Expressed MicroRNAs of Dairy Goat Mammary Gland Tissues Using Solexa Sequencing and Bioinformatics. PLoS ONE 2012, 7, e49463. [Google Scholar] [CrossRef]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Sci. Signal. 2003, 301, 336. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zeng, S.; Zhou, Z.W.; He, Z.X.; Zhou, S.F. Hsa-microRNA-181a is a regulator of a number of cancer genes and a biomarker for endometrial carcinoma in patients: A bioinformatic and clinical study and the therapeutic implication. Drug. Des. Devel. Ther. 2015, 9, 1103–1175. [Google Scholar]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, J.M.; Seila, A.C.; Yeo, G.W.; Sharp, P.A. RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18097–18102. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Zuker, M.; Jacobson, A. Using reliability information to annotate RNA secondary structures. RNA 1998, 4, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ruby, J.G.; Stark, A.; Johnston, W.K.; Kellis, M.; Bartel, D.P.; Lai, E.C. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007, 17, 1850–1864. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Egg | Larvae | Adult | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unique sRNAs | Percent (%) | Total sRNAs | Percent (%) | Unique sRNAs | Percent (%) | Total sRNAs | Percent (%) | Unique sRNAs | Percent (%) | Total sRNAs | Percent (%) | |
| Total | 4,908,986 | 100 | 18,162,337 | 100 | 1,506,602 | 100 | 8,090,736 | 100 | 2,913,657 | 100 | 11,807,326 | 100 |
| miRNA | 70,216 | 1.43 | 892,827 | 4.92 | 46,138 | 3.06 | 2,149,014 | 26.56 | 42,797 | 1.47 | 1,648,316 | 13.96 |
| rRNA | 64,751 | 1.32 | 1,665,040 | 9.17 | 43,714 | 2.90 | 575,299 | 7.11 | 30,408 | 1.04 | 484,924 | 4.11 |
| repeat | 165 | 0.00 | 285 | 0.00 | 52 | 0.00 | 92 | 0.00 | 87 | 0.00 | 231 | 0.00 |
| snRNA | 3713 | 0.08 | 20,631 | 0.11 | 1761 | 0.12 | 6378 | 0.08 | 2654 | 0.09 | 16,059 | 0.14 |
| snoRNA | 214 | 0.00 | 333 | 0.00 | 1077 | 0.07 | 1392 | 0.02 | 849 | 0.03 | 2142 | 0.02 |
| tRNA | 26,482 | 0.54 | 445,518 | 2.45 | 18,625 | 1.24 | 102,921 | 1.27 | 17,688 | 0.61 | 229,304 | 1.94 |
| unann | 4,743,445 | 96.63 | 15,137,703 | 83.35 | 1,395,235 | 92.61 | 5,255,640 | 64.96 | 2,819,174 | 96.76 | 9,426,350 | 79.83 |
| miRNA | Unique sRNAs Matched to miRNA Precursors | Total sRNAs Matched to miRNA Precursors | |
|---|---|---|---|
| Known miRNA in miRBase | 49 | —— | —— |
| Egg | 40 | 405 | 78,860 |
| Larvae | 44 | 517 | 1,439,717 |
| Adult | 43 | 523 | 746,431 |
| Novel miRNA | Mature Sequences (5′-3′) | Primer Sequences | Development Stages | Novel miRNA | Mature Sequences (5′-3′) | Primer Sequences | Development Stages |
|---|---|---|---|---|---|---|---|
| m0001 | ACTCGAGCTGCCCGTGCAAAAC | ACTCGAGCTGCCCGTG | Larva, adult | m0017 | TAAGTTAATCTCCAAGCCCAAT | TAAGTTAATCTCCAAG | adult |
| m0002 | ATCATAAGGATATCATCAATATT | ATCATAAGGATATCAT | Larva | m0018 | CTGGTTTTCACAATGATCGTCC | CTGGTTTTCACAATGA | All stages |
| m0003 | TATACGTCCAAAGCACTGAGG | TATACGTCCAAAGCAC | Larva | m0019 | ACGTGCTGCATCAGGTGCTTGTGA | ACGTGCTGCATCAGGT | egg |
| m0004 | CCTCACTCAGTTTGGCTGTGG | CCTCACTCAGTTTGGC | Larva, adult | m0020 | GTCCGGAAAATCGGTCGGCGA | GTCCGGAAAATCGGTC | egg |
| m0005 | TTTCATGTGACTTTTGAGGGC | TTTCATGTGACTTTTG | Larva, egg | m0021 | TGAGCATGGTTTTCGGCAACT | TGAGCATGGTTTTCGG | egg |
| m0006 | CCTTATCATTCGACTGTCCAGA | CCTTATCATTCGACTG | All stages | m0022 | TGAACCAACTGAACGACTGAA | TGAACCAACTGAACGA | egg |
| m0007 | TTGGGAAACAGAAGAGCGACGC | TTGGGAAACAGAAGAG | All stages | m0023 | GAGTGAAAGTAGGACGCCCA | GAGTGAAAGTAGGACG | egg |
| m0008 | GTGACTTCTCCGGTGCTGTGGA | GTGACTTCTCCGGTGC | Larva | m0024 | TAGTGGTTAGGATACCTGGCT | TAGTGGTTAGGATACC | egg |
| m0009 | AAAAATTGTGGTAGTGTCAAGC | AAAAATTGTGGTAGTG | Larva | m0025 | TATGTGGTATCGTTACAAGTG | TATGTGGTATCGTTAC | egg |
| m0010 | CTTCCCAAGCAGTTCCTGAAG | CTTCCCAAGCAGTTCC | Larva | m0026 | GGCCCGTTGGTCTAGGGGTAT | GGCCCGTTGGTCTAGG | egg |
| m0011 | AGGCATCTTTTGGAGTGCAAATG | AGGCATCTTTTGGAGT | adult | m0027 | TGGGCTAGTTGGTATGGCAT | TGGGCTAGTTGGTATG | egg |
| m0012 | TCGGATCCCATCCTCGTCGCCA | TCGGATCCCATCCTCG | adult | m0028 | TCGGACGGCATCAAGAAACGT | TCGGACGGCATCAAGA | egg |
| m0013 | TGGGCTTCCACGACGGCGGCAG | TGGGCTTCCACGACGG | adult | m0029 | TTATTTATTTAGTACATACT | TTATTTATTTAGTACA | egg |
| m0014 | GCGGAGCATTCGCGGTTGGCGGA | GCGGAGCATTCGCGGT | Adult, egg | m0030 | TTGACAAGCAACTATGTATCA | TTGACAAGCAACTATG | egg |
| m0015 | TCGAATCCTGTCGACTGCGCCA | TCGAATCCTGTCGACT | adult | m0031 | GATGAGTGTGGATCTAGTCATG | GATGAGTGTGGATCTA | egg |
| m0016 | TCCTGACCAAATGAGTGATGAGCA | TCCTGACCAAATGAGT | adult |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Zhao, S.; Ren, Q.; Wang, Q.; Chen, Z.; Cui, J.; Jing, Y.; Liu, P.; Yan, R.; Song, X.; et al. Dynamic Analysis of microRNAs from Different Life Stages of Rhipicephalus microplus (Acari: Ixodidae) by High-Throughput Sequencing. Pathogens 2022, 11, 1148. https://doi.org/10.3390/pathogens11101148
Luo J, Zhao S, Ren Q, Wang Q, Chen Z, Cui J, Jing Y, Liu P, Yan R, Song X, et al. Dynamic Analysis of microRNAs from Different Life Stages of Rhipicephalus microplus (Acari: Ixodidae) by High-Throughput Sequencing. Pathogens. 2022; 11(10):1148. https://doi.org/10.3390/pathogens11101148
Chicago/Turabian StyleLuo, Jin, Shuaiyang Zhao, Qiaoyun Ren, Qilin Wang, Zeyu Chen, Jingjing Cui, Yujiao Jing, Peiwen Liu, Ruofeng Yan, Xiaokai Song, and et al. 2022. "Dynamic Analysis of microRNAs from Different Life Stages of Rhipicephalus microplus (Acari: Ixodidae) by High-Throughput Sequencing" Pathogens 11, no. 10: 1148. https://doi.org/10.3390/pathogens11101148
APA StyleLuo, J., Zhao, S., Ren, Q., Wang, Q., Chen, Z., Cui, J., Jing, Y., Liu, P., Yan, R., Song, X., Liu, G., & Li, X. (2022). Dynamic Analysis of microRNAs from Different Life Stages of Rhipicephalus microplus (Acari: Ixodidae) by High-Throughput Sequencing. Pathogens, 11(10), 1148. https://doi.org/10.3390/pathogens11101148