
Next Generation Sequencing and Comparative Genomic Analysis Reveal Extreme Plasticity of Two Burkholderia glumae Strains HN1 and HN2
 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Isolation, DNA Extraction, and Sequencing
2.2. Genome Assembly and Annotation
2.3. Comparative Analysis against Sequenced and Annotated B. glumae Genomes
2.4. Positive Selection and Functional Enrichment Analysis
3. Results
3.1. Genome Assembly and Properties
3.2. Genome Annotation
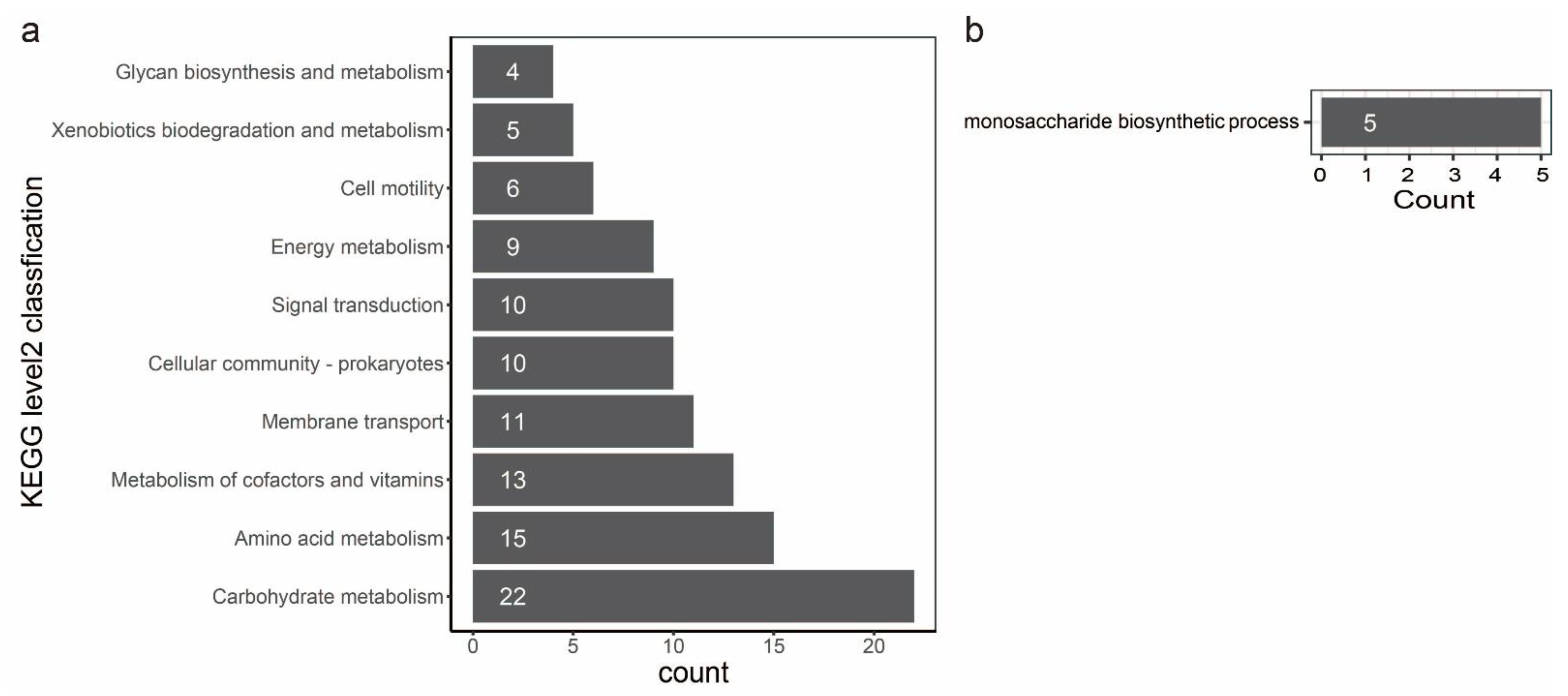
3.3. Pathway Analysis
3.4. EggNOG Functional Annotation
3.5. GO and Pfam Annotation
3.6. Enzymes Involved in Carbohydrate Metabolism
3.7. Virulence Factor Annotation
3.8. Genomic Island and Horizontal Gene Transfer
3.9. Genome Comparison
3.10. Positive Selection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ham, J.H.; Melanson, R.A.; Rush, M.C. Burkholderia glumae: Next major pathogen of rice? Mol. Plant Pathol. 2011, 12, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Zhou-Qi, C.; Bo, Z.; Guan-Lin, X.; Bin, L.; Shi-Wen, H. Research Status and Prospect of Burkholderia glumae, the Pathogen Causing Bacterial Panicle Blight. Rice Sci. 2016, 23, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Goto, K.; Ohata, K. New bacterial diseases of rice-bacterial (brown stripe and bacterial grain rot). Ann. Phytopathol. Soc. Jpn. 1956, 21, 46–47. [Google Scholar]
- Chien, C.C.; Chang, Y. The susceptibility of rice plants at different growth stages and of 21 commercial rice varieties to Pseudomonas glumae. J. Agric. Res. China 1987, 36, 302–310. [Google Scholar]
- Jeong, Y.; Kim, J.; Kim, S.; Kang, Y.; Nagamatsu, T.; Hwang, I. Toxoflavin Produced by Burkholderia glumae Causing Rice Grain Rot Is Responsible for Inducing Bacterial Wilt in Many Field Crops. Plant Dis. 2003, 87, 890–895. [Google Scholar] [CrossRef] [Green Version]
- Zeigler, R.; Alvarez, E. Grain discoloration of rice caused by Pseudomonas glumae in Latin America. Plant Dis. 1989, 73, 368. [Google Scholar] [CrossRef]
- Trung, H.M.; Van Van, N.; Vien, N.V.; Lien, M. Occurrence of rice grain rot disease in Viet Nam. Int. Rice Res. Notes 1993, 18, 30. [Google Scholar]
- Mondal, K.K.; Mani, C.; Verma, G. Emergence of Bacterial Panicle Blight Caused by Burkholderia glumae in North India. Plant Dis. 2015, 99, 1268. [Google Scholar] [CrossRef]
- Zhou, X.; McClung, A.; Way, M.; Jo, Y.; Tabien, R.; Wilson, L. Severe outbreak of bacterial panicle blight across Texas Rice Belt in 2010. Phytopathology 2011, 101, S205. [Google Scholar]
- Shew, A.M.; Durand-Morat, A.; Nalley, L.L.; Zhou, X.-G.; Rojas, C.; Thoma, G. Warming increases bacterial panicle blight (Burkholderia glumae) occurrences and impacts on USA rice production. PLoS ONE 2019, 14, e0219199. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, R.; Shahjahan, A.K.M.; Yuan, X.L.; Dickstein, E.R.; Groth, D.E.; Clark, C.A.; Cartwright, R.D.; Rush, M.C. Burkholderia glumae and B. gladioli cause bacterial panicle blight in rice in the southern United States. Plant Dis. 2009, 93, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahjahan, A.; Rush, M.; Groth, D.; Clark, C. Panicle blight. Rice J. 2000, 15, 26–29. [Google Scholar]
- Kurita, T. A few studies on factors associated with infection of bacterial grain rot of rice. Ann. Phytopathol. Soc. Jpn. 1964, 29, 60. [Google Scholar]
- Sato, Z.; Koiso, Y.; Iwasaki, S.; Matsuda, I.; Shirata, A. Toxins produced by Pseudomonas glumae. Jpn. J. Phytopathol. 1989, 55, 353–356. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.G.; Kang, Y.; Jang, J.Y.; Jog, G.J.; Lim, J.Y.; Kim, S.; Suga, H.; Nagamatsu, T.; Hwang, I. Quorum sensing and the LysR-type transcriptional activator ToxR regulate toxoflavin biosynthesis and transport in Burkholderia glumae. Mol. Microbiol. 2004, 54, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Sawada, H.; Azegami, K.; Tsuchiya, K. Molecular characterization of the tox operon involved in toxoflavin biosynthesis of Burkholderia glumae. J. Gen. Plant Pathol. 2004, 70, 97–107. [Google Scholar] [CrossRef]
- Devescovi, G.; Bigirimana, J.; Degrassi, G.; Cabrio, L.; LiPuma, J.J.; Kim, J.; Hwang, I.; Venturi, V. Involvement of a quorum-sensing-regulated lipase secreted by a clinical isolate of Burkholderia glumae in severe disease symptoms in rice. Appl. Environ. Microbiol. 2007, 73, 4950–4958. [Google Scholar] [CrossRef] [Green Version]
- Frenken, L.G.; Egmond, M.R.; Batenburg, A.M.; Verrips, C.T. Pseudomonas glumae lipase: Increased proteolytic stabifity by protein engineering. Protein Eng. Des. Sel. 1993, 6, 637–642. [Google Scholar] [CrossRef]
- El Khattabi, M.; Van Gelder, P.; Bitter, W.; Tommassen, J. Role of the lipase-specific foldase of Burkholderia glumae as a steric chaperone. J. Biol. Chem. 2000, 275, 26885–26891. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Choi, O.; Jeong, Y.; Jeong, J.-E.; Lim, J.Y.; Kim, M.; Moon, J.S.; Suga, H.; Hwang, I. Regulation of polar flagellum genes is mediated by quorum sensing and FlhDC in Burkholderia glumae. Mol. Microbiol. 2007, 64, 165–179. [Google Scholar] [CrossRef]
- Lee, C.; Mannaa, M.; Kim, N.; Kim, J.; Choi, Y.; Kim, S.H.; Jung, B.; Lee, H.-H.; Lee, J.; Seo, Y.-S. Stress Tolerance and Virulence-Related Roles of Lipopolysaccharide in Burkholderia glumae. Plant Pathol. J. 2019, 35, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mannaa, M.; Kim, N.; Lee, C.; Kim, J.; Park, J.; Lee, H.-H.; Seo, Y.-S. The Roles of Two hfq Genes in the Virulence and Stress Resistance of Burkholderia glumae. Plant Pathol. J. 2018, 34, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Degrassi, G.; Devescovi, G.; Kim, J.; Hwang, I.; Venturi, V. Identification, characterization and regulation of two secreted polygalacturonases of the emerging rice pathogen Burkholderia glumae. FEMS Microbiol. Ecol. 2008, 65, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Choi, O.; Goo, E.; Kim, N.; Kim, H.; Kang, Y.; Kim, J.; Moon, J.S.; Hwang, I. The quorum sensing-dependent gene katG of Burkholderia glumae is important for protection from visible light. J. Bacteriol. 2009, 191, 4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Kim, J.; Kim, S.; Kim, H.; Lim, J.Y.; Kim, M.; Kwak, J.; Moon, J.S.; Hwang, I. Proteomic analysis of the proteins regulated by HrpB from the plant pathogenic bacterium Burkholderia glumae. Proteomics 2008, 8, 106–121. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Silva, N.D.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Galore, T. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 September 2022).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. Island Viewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Kosoy, M.Y.; Dittmar, K. HGTector: An automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 2014, 15, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2015, 8, 12–24. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2021, 50, D801–D807. [Google Scholar] [CrossRef]
- Harris, S.R. SKA: Split kmer analysis toolkit for bacterial genomic epidemiology. Biorxiv 2018, 453142. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2017, 34, 292–293. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Snipen, L.-G.; Liland, K.H. micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015, 16, 79. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019, 47, 5539–5549. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2019, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Rendón, J.; Rada-Bravo, A.M.; Cárdenas-Brito, S.; Corredor, M.; Restrepo-Pineda, E.; Benítez-Páez, A. Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genom. 2016, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Hotopp, J.C.D.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Dobrindt, U.; Zdziarski, J.; Salvador, E.; Hacker, J. Bacterial genome plasticity and its impact on adaptation during persistent infection. Int. J. Med. Microbiol. 2010, 300, 363–366. [Google Scholar] [CrossRef]
- Harrison, E.; Brockhurst, M.A. Plasmid-mediated horizontal gene transfer is a coevolutionary process. Trends Microbiol. 2012, 20, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [Green Version]
- Francis, F.; Kim, J.; Ramaraj, T.; Farmer, A.; Rush, M.C.; Ham, J.H. Comparative genomic analysis of two Burkholderia glumae strains from different geographic origins reveals a high degree of plasticity in genome structure associated with genomic islands. Mol. Genet. Genom. 2013, 288, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, J.J.; Kim, I.; Mannaa, M.; Park, J.; Kim, J.; Lee, H.; Lee, S.; Park, D.; Sul, W.J.; et al. Type VI secretion systems of plant-pathogenic Burkholderia glumae BGR1 play a functionally distinct role in interspecies interactions and virulence. Mol. Plant Pathol. 2020, 21, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.; Lee, J.; Shin, D.; Park, D.-S.; Lim, J.-S.; Choi, I.-Y.; Seo, Y.-S. Understanding pathogenic Burkholderia glumae metabolic and signaling pathways within rice tissues through in vivo transcriptome analyses. Gene 2014, 547, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, P.-H.; Nie, W.-H.; Cui, Z.-Q.; Li, H.-Y.; Wu, Y.; Yiming, A.; Fu, L.-Y.; Ahmad, I.; Chen, G.-Y.; et al. Horizontal gene transfer of a syp homolog contributes to the virulence of Burkholderia glumae. J. Integr. Agric. 2021, 20, 3222–3229. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HN1 | HN2 | |
|---|---|---|
| Status | Complete | Complete |
| Genome size (bp) | 6,680,415 | 6,560,085 |
| GC content (%) | 68.06 | 68.34 |
| DNA replicons | 5 | 3 |
| Total genes | 5867 | 5717 |
| Protein-coding genes | 5434 | 5278 |
| RNA genes | 82 | 82 |
| rRNA genes | 15 | 15 |
| tRNA genes | 63 | 63 |
| ncRNA | 4 | 4 |
| Pseudogenes | 351 | 357 |
| CRISPR | 1 | 2 |
| Pathogenic associated genes | 195 | 121 |
| Genes with function prediction | 5045 | 4975 |
| Genes connected to KEGG pathways | 2487 | 2477 |
| Genes connected to KEGG Orthology (KO) | 2955 | 2930 |
| Genes assigned to COGs | 4604 | 4545 |
| Genes assigned to GOs | 3420 | 2812 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Nie, W.; Yiming, A.; Wang, P.; Wu, Y.; Huang, J.; Ahmad, I.; Chen, G.; Guo, L.; Zhu, B. Next Generation Sequencing and Comparative Genomic Analysis Reveal Extreme Plasticity of Two Burkholderia glumae Strains HN1 and HN2. Pathogens 2022, 11, 1265. https://doi.org/10.3390/pathogens11111265
Wang S, Nie W, Yiming A, Wang P, Wu Y, Huang J, Ahmad I, Chen G, Guo L, Zhu B. Next Generation Sequencing and Comparative Genomic Analysis Reveal Extreme Plasticity of Two Burkholderia glumae Strains HN1 and HN2. Pathogens. 2022; 11(11):1265. https://doi.org/10.3390/pathogens11111265
Chicago/Turabian StyleWang, Sai, Wenhan Nie, Ayizekeranmu Yiming, Peihong Wang, Yan Wu, Jin Huang, Iftikhar Ahmad, Gongyou Chen, Longbiao Guo, and Bo Zhu. 2022. "Next Generation Sequencing and Comparative Genomic Analysis Reveal Extreme Plasticity of Two Burkholderia glumae Strains HN1 and HN2" Pathogens 11, no. 11: 1265. https://doi.org/10.3390/pathogens11111265
APA StyleWang, S., Nie, W., Yiming, A., Wang, P., Wu, Y., Huang, J., Ahmad, I., Chen, G., Guo, L., & Zhu, B. (2022). Next Generation Sequencing and Comparative Genomic Analysis Reveal Extreme Plasticity of Two Burkholderia glumae Strains HN1 and HN2. Pathogens, 11(11), 1265. https://doi.org/10.3390/pathogens11111265