
An Exploratory Study on the Microbiome of Northern and Southern Populations of Ixodes scapularis Ticks Predicts Changes and Unique Bacterial Interactions

 , , ,
, , ,  and
and 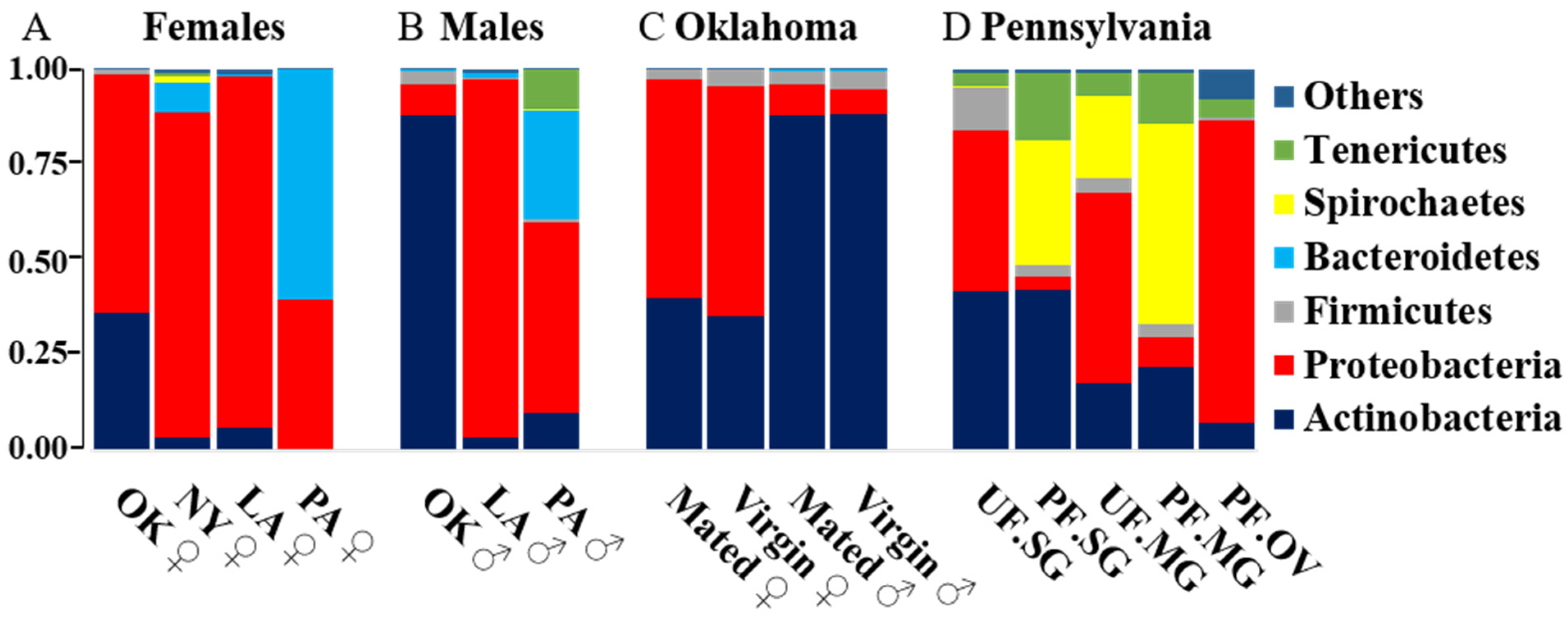{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Illumina MiSeq 16S V1–V3 Sequencing
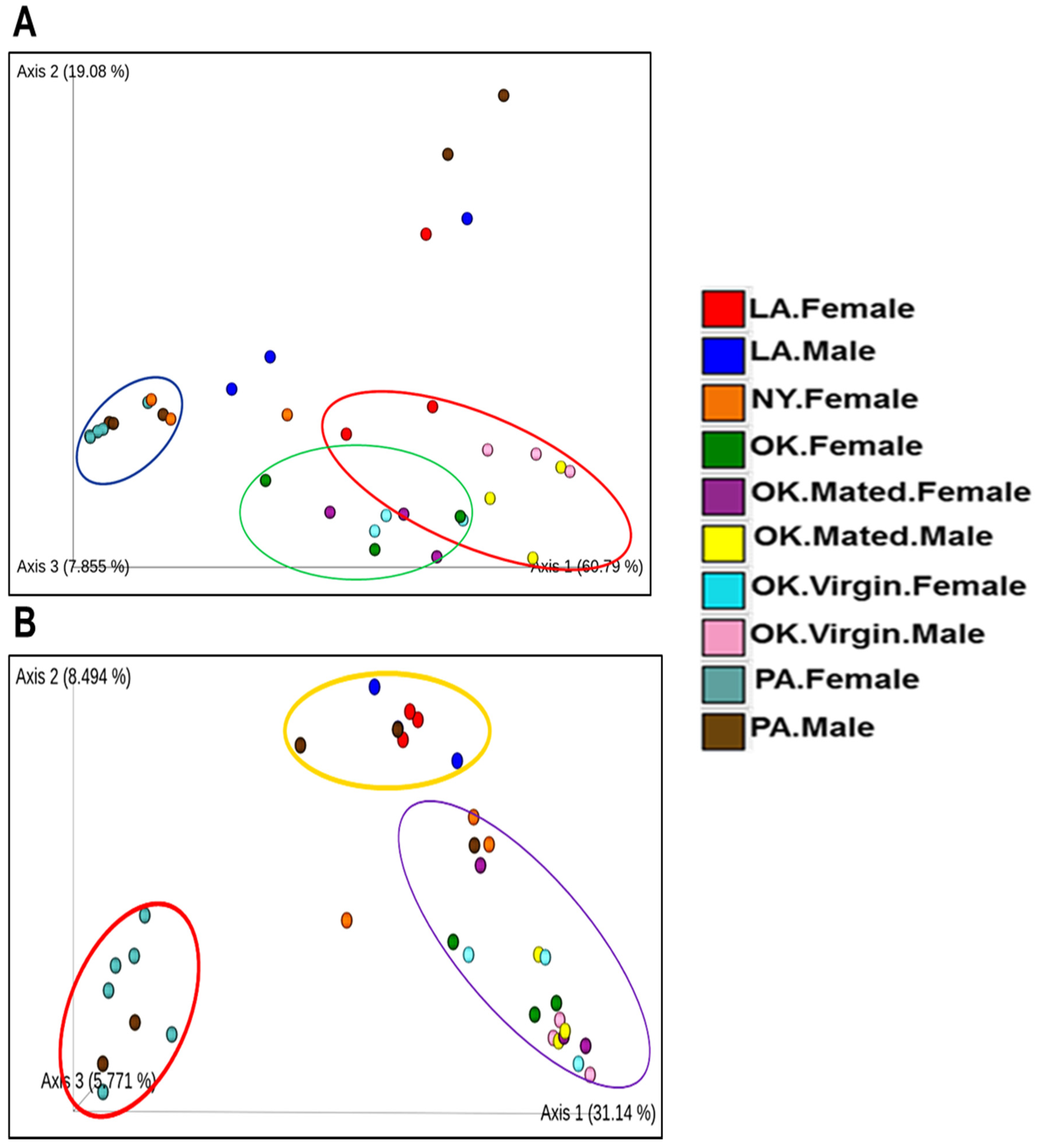
2.2. Comparative Analysis of Bacterial Communities Residing within Ix. scapularis
2.3. Bacterial Composition of Unfed and Partially Engorged Tick Tissues from Pennsylvania (PA)
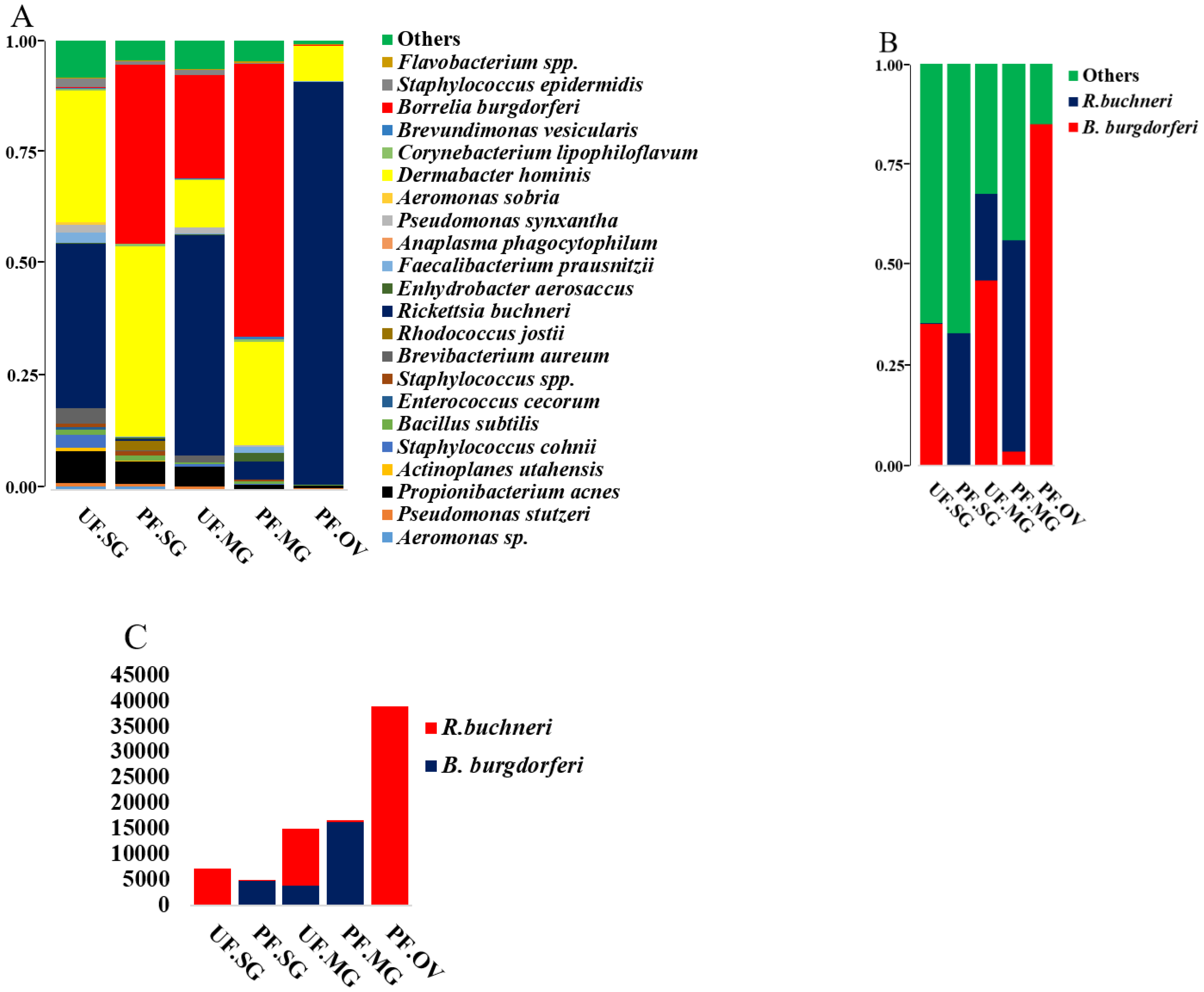
2.4. Presence of Rickettsia Buchneri in Partially Fed Tick Tissues and Pattern of Likely Competitive Interactions with B. burgdorferi s.l.
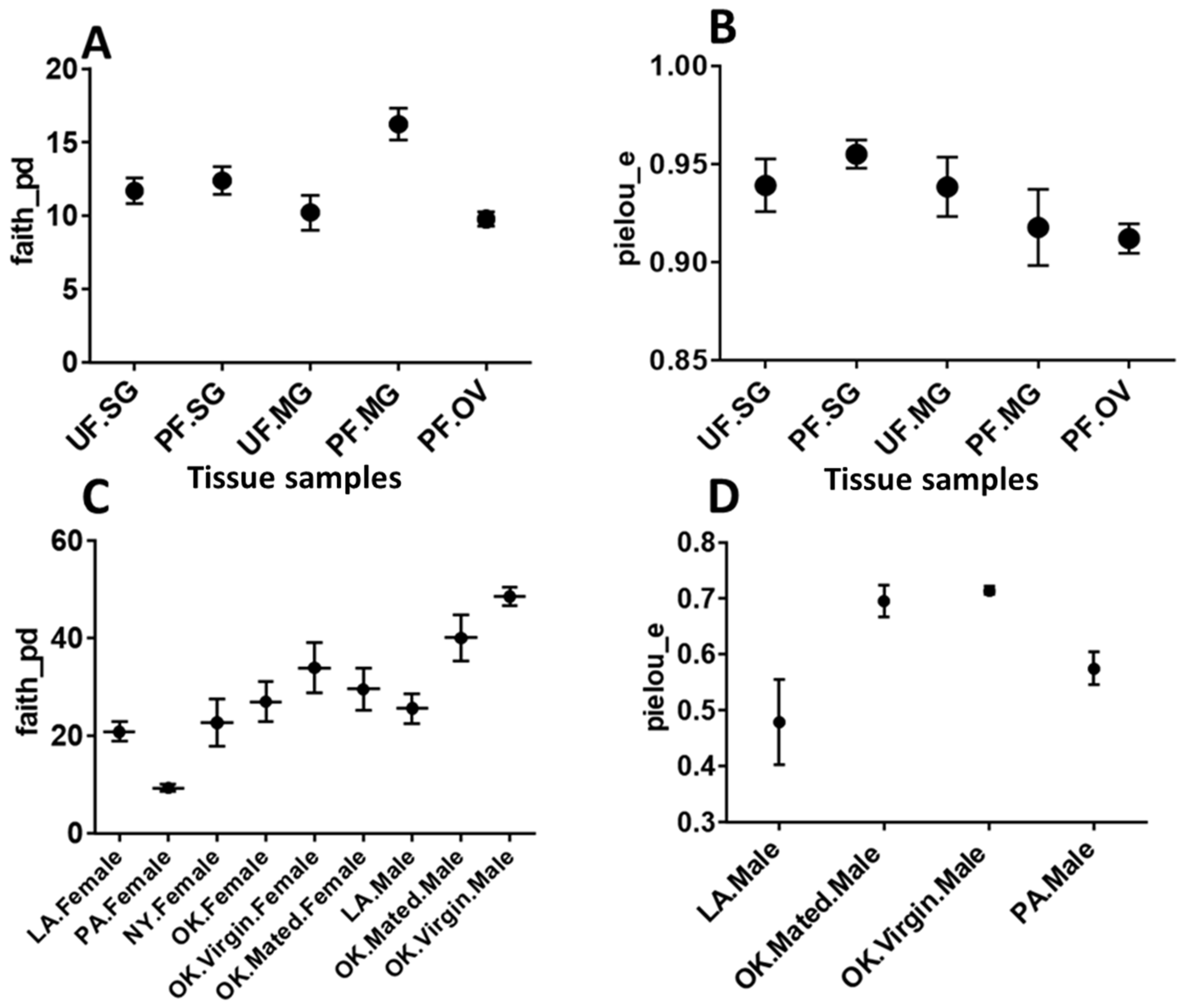
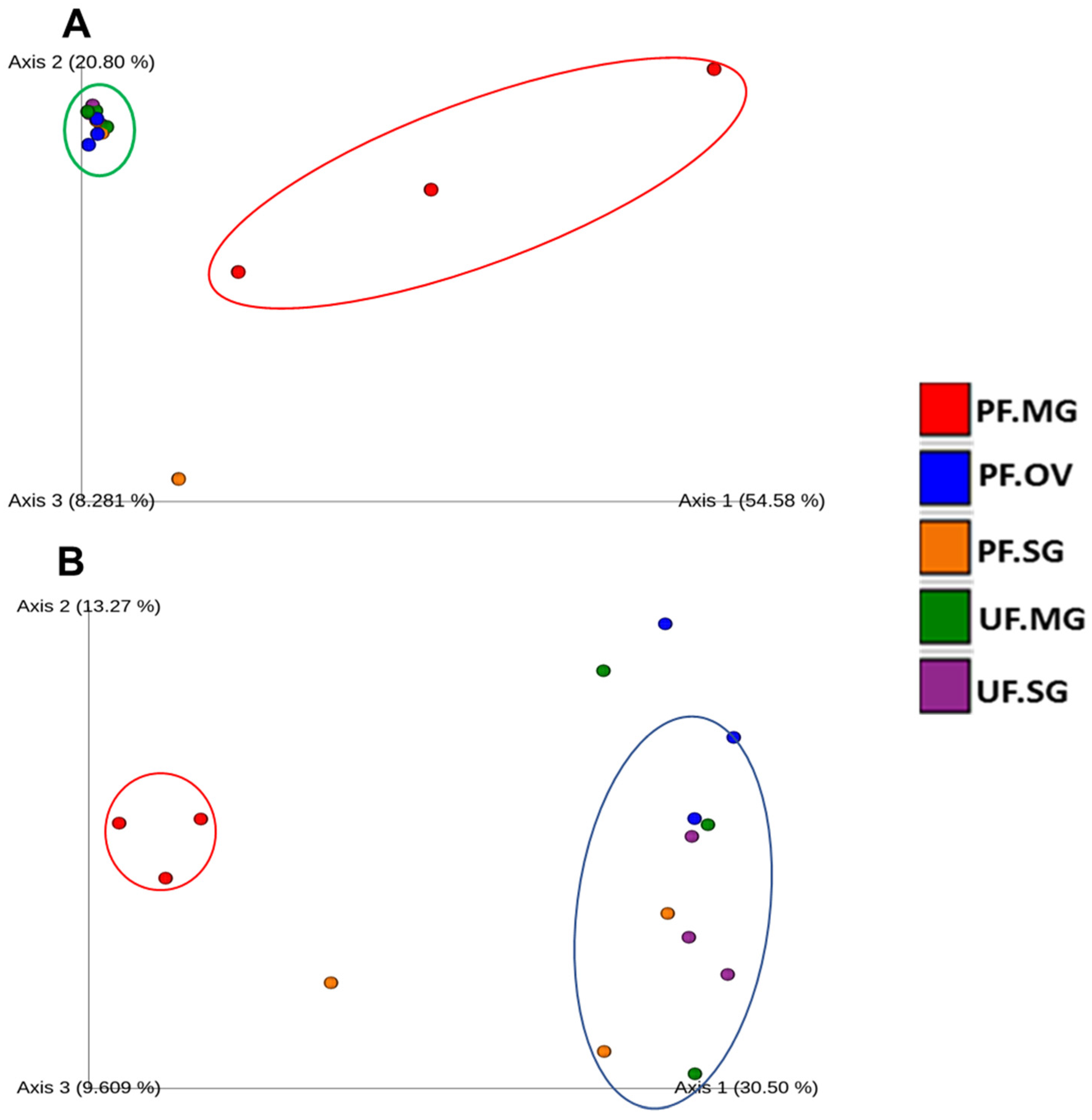
2.5. Diversity Analysis
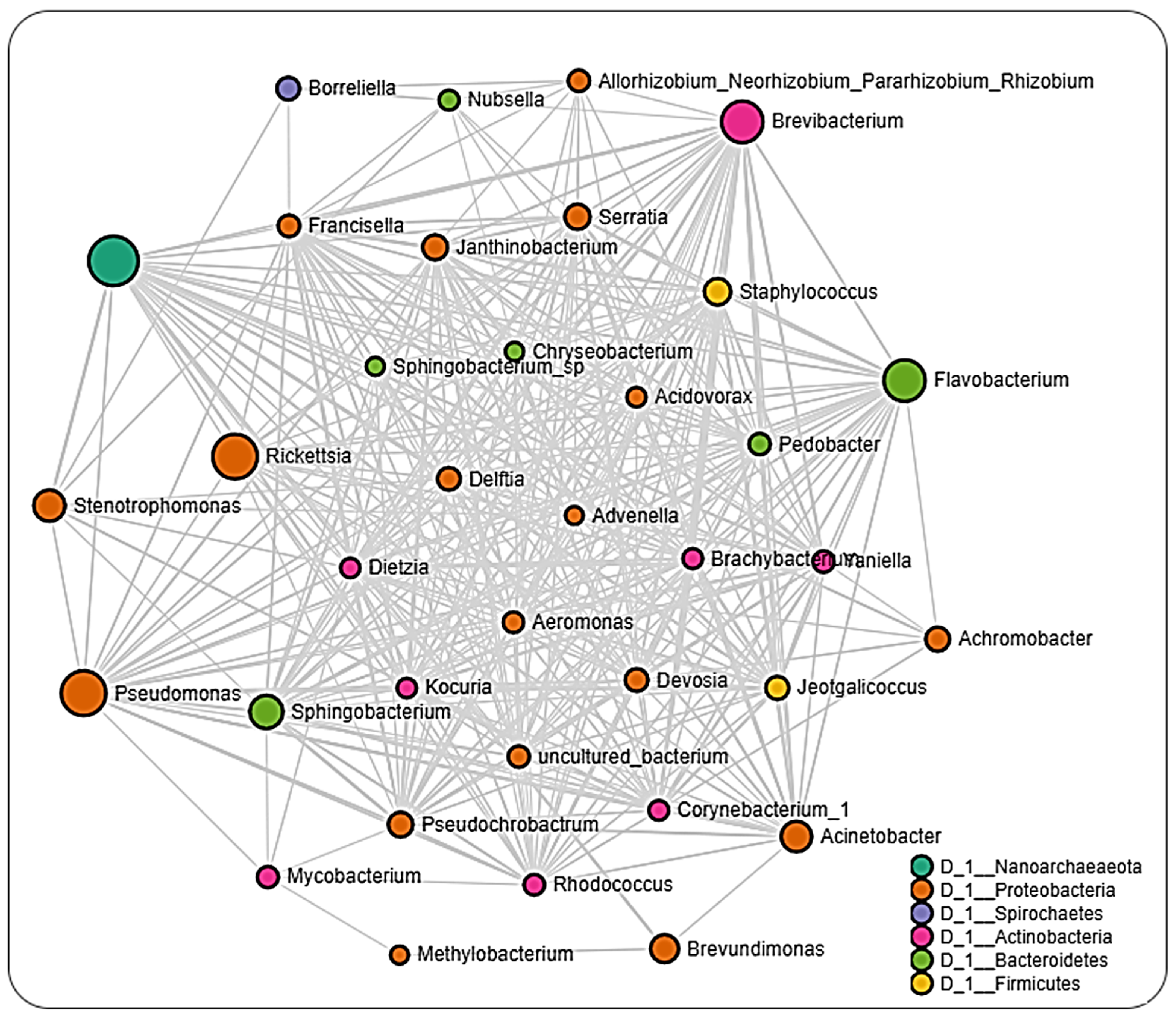
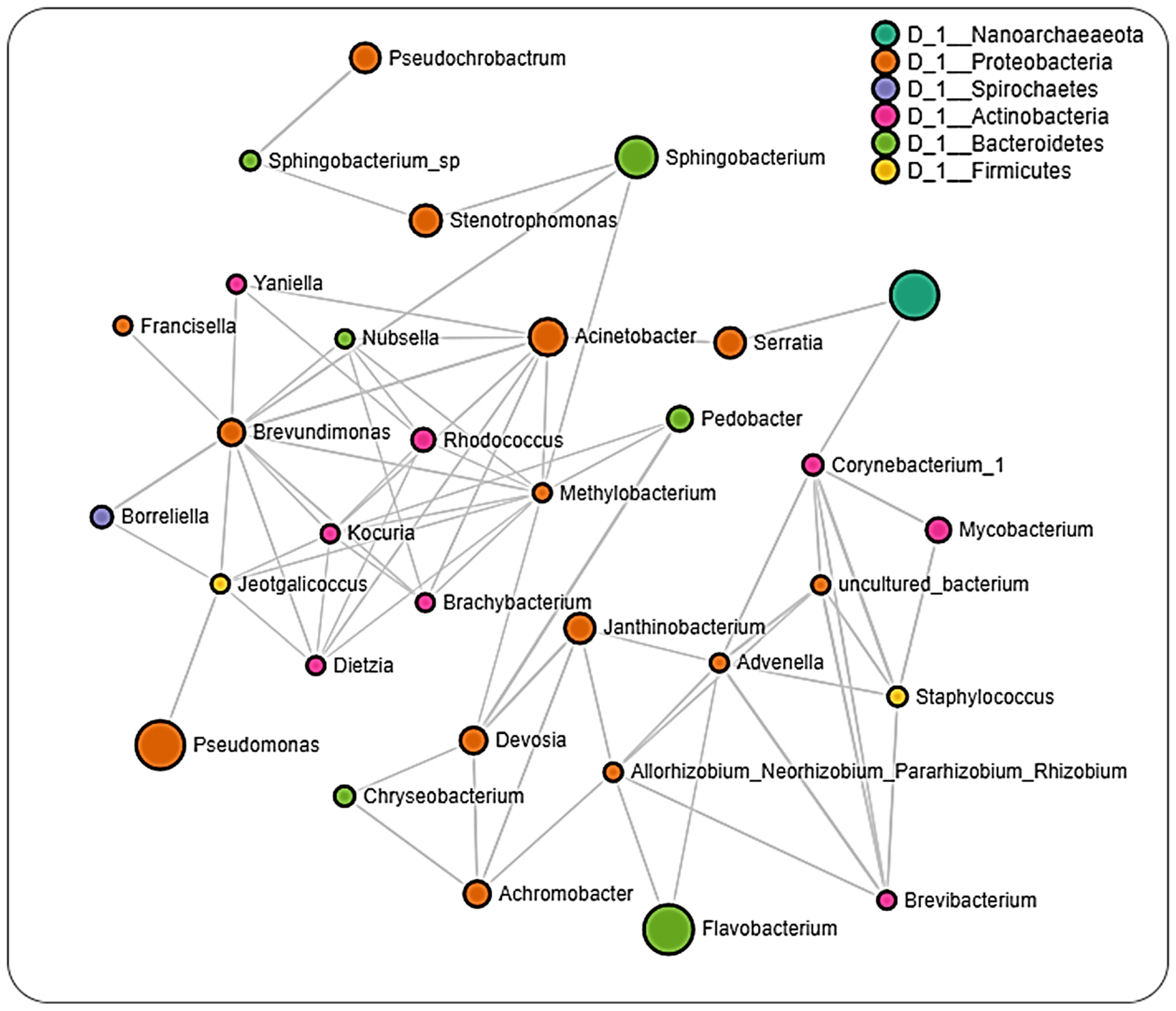
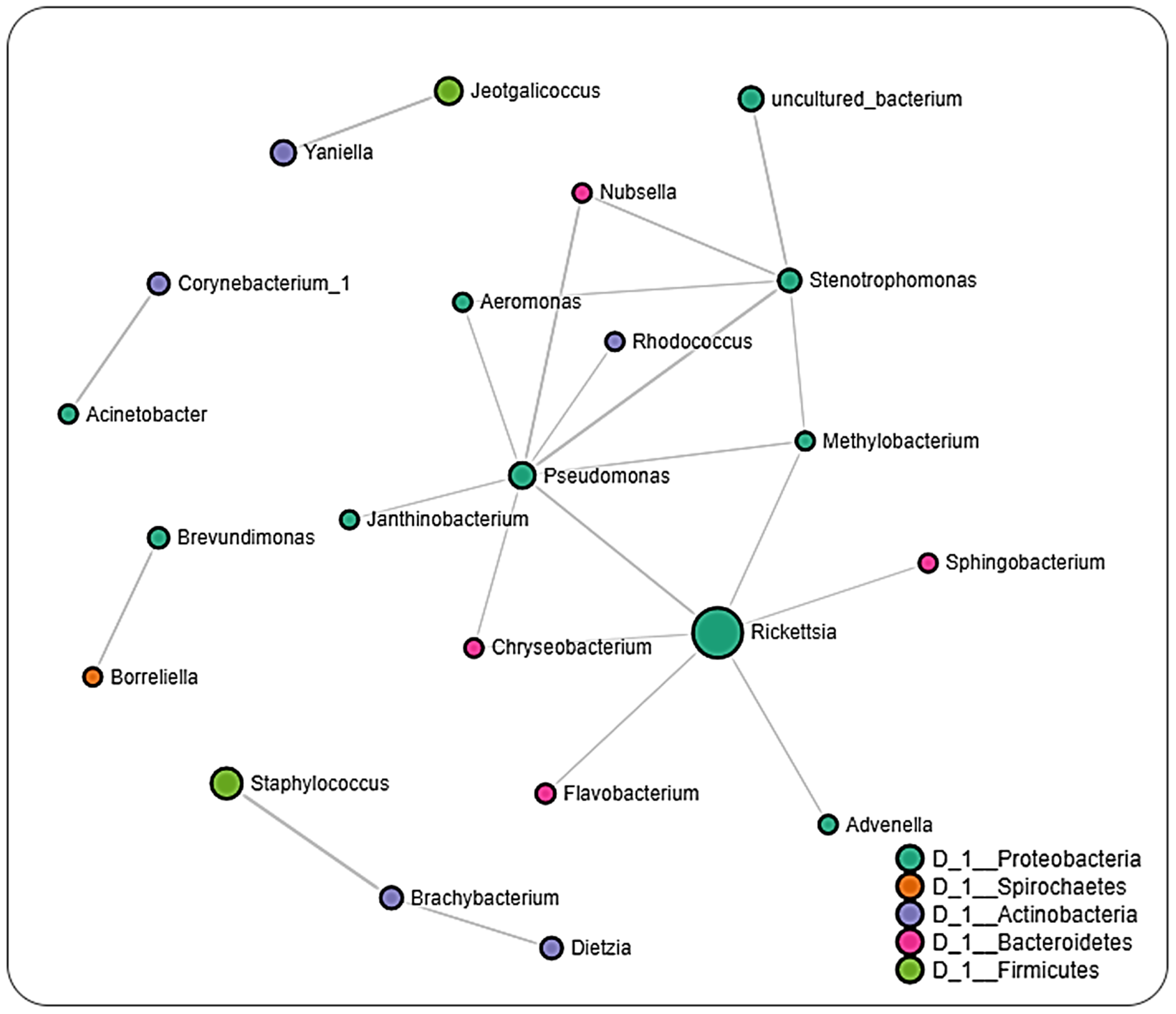
2.6. Microbial Interactions
3. Discussion
4. Materials and Methods
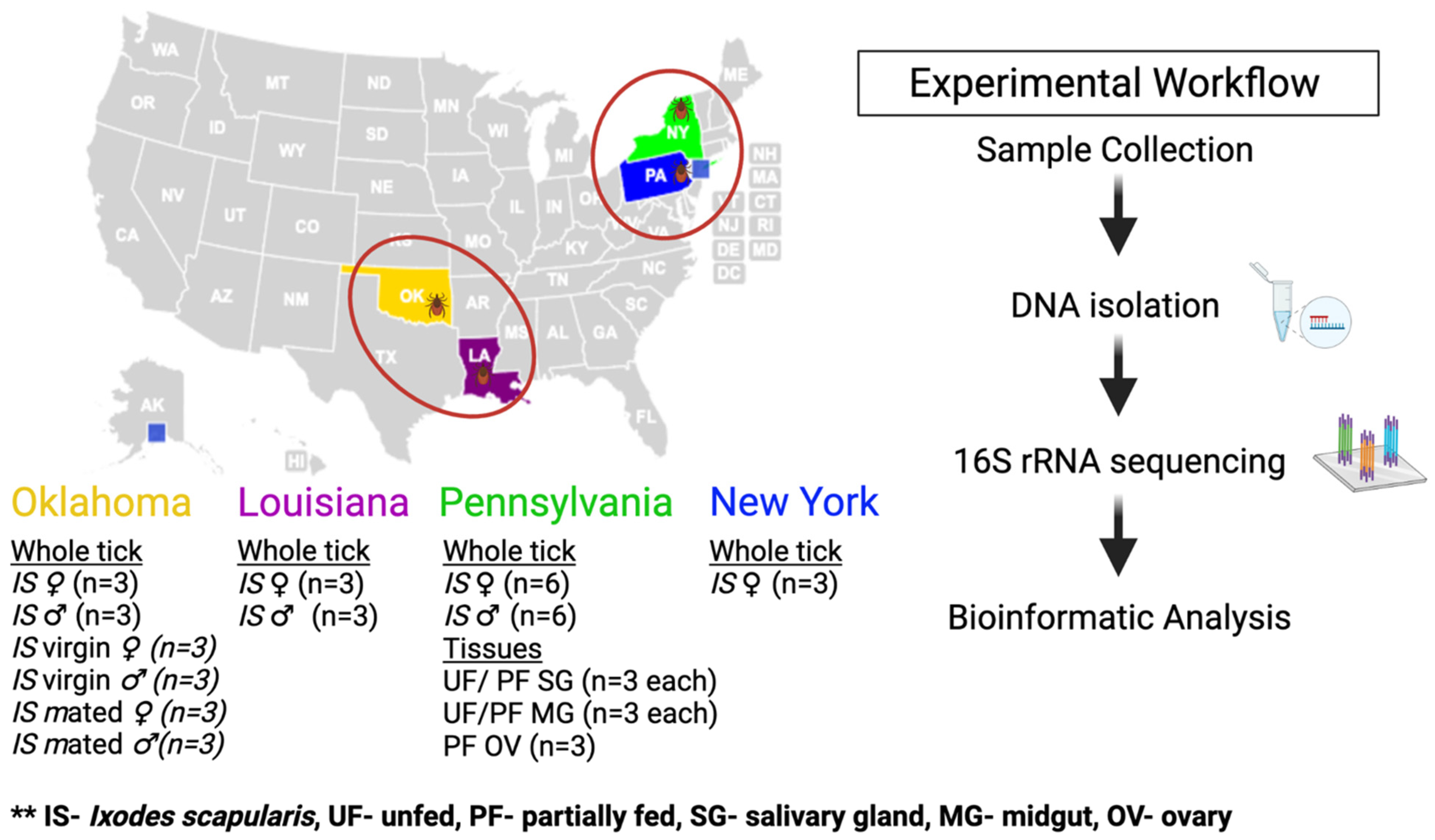
4.1. Ticks
4.2. DNA Isolation
4.3. Illumina Library Preparation and 16S rRNA Sequencing
4.4. Data Processing
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvell, C.D.; Mitchell, C.E.; Ward, J.R.; Altizer, S.; Dobson, A.P.; Ostfeld, R.S.; Samuel, M.D. Climate warming and disease risks for terrestrial and marine biota. Science 2002, 296, 2158–2162. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.A.; Grace, D.; Kock, R.; Alonso, S.; Rushton, J.; Said, M.Y.; McKeever, D.; Mutua, F.; Young, J.; McDermott, J.; et al. Zoonosis emergence linked to agricultural intensification and environmental change. Proc. Natl. Acad. Sci. USA 2013, 110, 8399–8404. [Google Scholar] [CrossRef] [Green Version]
- Keesing, F.; Belden, L.K.; Daszak, P.; Dobson, A.; Harvell, C.D.; Holt, R.D.; Hudson, P.; Jolles, A.; Jones, K.E.; Mitchell, C.E.; et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 2010, 468, 647–652. [Google Scholar] [CrossRef]
- Kugeler, K.J.; Farley, G.M.; Forrester, J.D.; Mead, P.S. Geographic Distribution and Expansion of Human Lyme Disease, United States. Emerg. Infect. Dis. 2015, 21, 1455–1457. [Google Scholar] [CrossRef] [PubMed]
- Radolf, J.D.; Caimano, M.J.; Stevenson, B.; Hu, L.T. Of ticks, mice and men: Understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat. Rev. Microbiol. 2012, 10, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinckley, A.F.; Connally, N.P.; Meek, J.I.; Johnson, B.J.; Kemperman, M.M.; Feldman, K.A.; White, J.L.; Mead, P.S. Lyme disease testing by large commercial laboratories in the United States. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2014, 59, 676–681. [Google Scholar] [CrossRef]
- Roy-Dufresne, E.; Logan, T.; Simon, J.A.; Chmura, G.L.; Millien, V. Poleward expansion of the white-footed mouse (Peromyscus leucopus) under climate change: Implications for the spread of lyme disease. PLoS ONE 2013, 8, e80724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, C.X.; Moy, F.; Daniels, T.J.; Godfrey, H.P.; Cabello, F.C. Molecular analysis of microbial communities identified in different developmental stages of Ixodes scapularis ticks from Westchester and Dutchess Counties, New York. Environ. Microbiol. 2006, 8, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Ostfeld, R.S.; Levi, T.; Keesing, F.; Oggenfuss, K.; Canham, C.D. Tick-borne disease risk in a forest food web. Ecology 2018, 99, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Adelson, M.E.; Rao, R.V.; Tilton, R.C.; Cabets, K.; Eskow, E.; Fein, L.; Occi, J.L.; Mordechai, E. Prevalence of Borrelia burgdorferi, Bartonella spp., Babesia microti, and Anaplasma phagocytophila in Ixodes scapularis ticks collected in Northern New Jersey. J. Clin. Microbiol. 2004, 42, 2799–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, L.; Eisen, R.J.; Mun, J.; Salkeld, D.J.; Lane, R.S. Transmission cycles of Borrelia burgdorferi and B. bissettii in relation to habitat type in northwestern California. J. Vector Ecol. J. Soc. Vector Ecol. 2009, 34, 81–91. [Google Scholar] [CrossRef]
- Maggi, R.G.; Reichelt, S.; Toliver, M.; Engber, B. Borrelia species in Ixodes affinis and Ixodes scapularis ticks collected from the coastal plain of North Carolina. Ticks Tick-Borne Dis. 2010, 1, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Oliver, J.H.; James, A.M., Jr.; Durden, L.A.; Banks, C.W. Prevalence of borrelia burgdorferi sensu lato infection among rodents and host-seeking ticks in South Carolina. J. Med. Entomol. 2002, 39, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; Andreadis, T.G.; Falco, R.C.; Ziegler, L.B.; Hathaway, N.; et al. Variation in the Microbiota of Ixodes Ticks with Regard to Geography, Species, and Sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef] [Green Version]
- Clay, K.; Klyachko, O.; Grindle, N.; Civitello, D.; Oleske, D.; Fuqua, C. Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol. Ecol. 2008, 17, 4371–4381. [Google Scholar] [CrossRef] [PubMed]
- Carpi, G.; Cagnacci, F.; Wittekindt, N.E.; Zhao, F.; Qi, J.; Tomsho, L.P.; Drautz, D.I.; Rizzoli, A.; Schuster, S.C. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS ONE 2011, 6, e25604. [Google Scholar] [CrossRef]
- Van Overbeek, L.; Gassner, F.; van der Plas, C.L.; Kastelein, P.; Nunes-da Rocha, U.; Takken, W. Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol. Ecol. 2008, 66, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Clay, K.; Fuqua, C. The tick microbiome: Diversity, distribution and influence of the internal microbial community for a blood-feeding disease vector. In Critical Needs and Gaps in Understanding Prevention, Amelioration, and Resolution of Lyme and Other Tick-Borne Diseases: The Short-Term and Long-Term Outcomes. Workshop Report; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Nakao, R.; Abe, T.; Nijhof, A.M.; Yamamoto, S.; Jongejan, F.; Ikemura, T.; Sugimoto, C. A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks. ISME J. 2013, 5, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Budachetri, K.; Browning, R.E.; Adamson, S.W.; Dowd, S.E.; Chao, C.C.; Ching, W.M.; Karim, S. An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J. Med. Entomol. 2014, 51, 119–129. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Gonzalez, A.; Van Treuren, W.; Weiss, S.; Parobek, C.M.; Juliano, J.J.; Knight, R.; Roe, R.M.; Apperson, C.S.; Meshnick, S.R. Diversity of Rickettsiales in the microbiome of the lone star tick, Amblyomma americanum. Appl. Environ. Microbiol. 2014, 80, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Landesman, W.J.; Mulder, K.; Allan, B.F.; Bashor, L.A.; Keesing, F.; LoGiudice, K.; Ostfeld, R.S. Potential effects of blood meal host on bacterial community composition in Ixodes scapularis nymphs. Ticks Tick-Borne Dis. 2019, 10, 523–527. [Google Scholar] [CrossRef]
- Martin, P.A.; Schmidtmann, E.T. Isolation of aerobic microbes from Ixodes scapularis (Acari: Ixodidae), the vector of Lyme disease in the eastern United States. J. Econ. Entomol. 1998, 91, 864–868. [Google Scholar] [CrossRef]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Ahantarig, A.; Trinachartvanit, W.; Baimai, V.; Grubhoffer, L. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol. 2013, 58, 419–428. [Google Scholar] [CrossRef]
- Gall, C.A.; Reif, K.E.; Scoles, G.A.; Mason, K.L.; Mousel, M.; Noh, S.M.; Brayton, K.A. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J. 2016, 10, 1846–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Jasinskas, A.; Barbour, A.G. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS ONE 2007, 2, e405. [Google Scholar] [CrossRef] [PubMed]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Clow, K.M.; Weese, J.S.; Rousseau, J.; Jardine, C.M. Microbiota of field-collected Ixodes scapularis and Dermacentor variabilis from eastern and southern Ontario, Canada. Ticks Tick-Borne Dis. 2018, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.Y.; Griggs, R.; Chicana, B.; Miller, C.; Swei, A. Vertical vs. horizontal transmission of the microbiome in a key disease vector, Ixodes pacificus. Mol. Ecol. 2017, 26, 6578–6589. [Google Scholar] [CrossRef]
- Thapa, S.; Zhang, Y.; Allen, M.S. Bacterial microbiomes of Ixodes scapularis ticks collected from Massachusetts and Texas, USA. BMC Microbiol. 2019, 19, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkerhoff, R.J.; Clark, C.; Ocasio, K.; Gauthier, D.T.; Hynes, W.L. Factors affecting the microbiome of Ixodes scapularis and Amblyomma americanum. PLoS ONE 2020, 15, e0232398. [Google Scholar] [CrossRef]
- Sperling, J.; Fitzgerald, D.; Sperling, F.; Magor, K.E. Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range. Trop. Med. Infect. Dis. 2020, 5, 173. [Google Scholar] [CrossRef] [PubMed]
- Hawlena, H.; Rynkiewicz, E.; Toh, E.; Alfred, A.; Durden, L.A.; Hastriter, M.W.; Nelson, D.E.; Rong, R.; Munro, D.; Dong, Q. The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J. 2013, 7, 221–223. [Google Scholar] [CrossRef] [Green Version]
- Williams-Newkirk, A.J.; Rowe, L.A.; Mixson-Hayden, T.R.; Dasch, G.A. Characterization of the bacterial communities of life stages of free living lone star ticks (Amblyomma americanum). PLoS ONE 2014, 9, e102130. [Google Scholar] [CrossRef]
- Zhang, X.C.; Yang, Z.N.; Lu, B.; Ma, X.F.; Zhang, C.X.; Xu, H.J. The composition and transmission of microbiome in hard tick, Ixodes persulcatus, during blood meal. Ticks Tick-Borne Dis. 2014, 5, 864–870. [Google Scholar] [CrossRef]
- Burtis, J.C.; Yavitt, J.B.; Fahey, T.J.; Ostfeld, R.S. Ticks as soildwelling arthropods: An intersection between disease and soil ecology. J. Med. Entomol. 2019, 56, 1555–1564. [Google Scholar] [CrossRef]
- Rynkiewicz, E.C.; Hemmerich, C.; Rusch, D.B.; Fuqua, C.; Clay, K. Concordance of bacterial communities of two tick species and blood of their shared rodent host. Mol. Ecol. 2015, 24, 2566–2579. [Google Scholar] [CrossRef]
- Oliver, J.D.; Price, L.D.; Burkhardt, N.Y.; Heu, C.C.; Khoo, B.S.; Thorpe, C.J.; Kurtti, T.J.; Munderloh, U.G. Growth Dynamics and Antibiotic Elimination of Symbiotic Rickettsia buchneri in the Tick Ixodes scapularis (Acari: Ixodidae). Appl. Environ. Microbiol. 2021, 87, e01672-20. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; Deponte, K.; Fish, D.; Fikrig, E. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Macaluso, K.R.; Sonenshine, D.E.; Ceraul, S.M.; Azad, A.F. Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J. Med. Entomol. 2002, 39, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Ross, B.D.; Hayes, B.; Radey, M.C.; Lee, X.; Josek, T.; Bjork, J.; Mougous, J.D. Ixodes scapularis does not harbor a stable midgut micro-biome. ISME J. 2018, 12, 2596–2607. [Google Scholar] [CrossRef] [Green Version]
- Simhadri, R.K.; Fast, E.M.; Guo, R.; Schultz, M.J.; Vaisman, N.; Ortiz, L.; Bybee, J.; Slatko, B.E.; Frydman, H.M. The Gut Commensal Microbiome of Drosophila melanogaster Is Modified by the Endosymbiont Wolbachia. mSphere 2017, 2, e00287-17. [Google Scholar] [CrossRef] [Green Version]
- Kocan, K.M.; de la Fuente, J.; Blouin, E.F.; Coetzee, J.F.; Ewing, S.A. The natural history of Anaplasma marginale. Vet. Parasitol. 2010, 167, 95–107. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Joardar, V.; Williams, K.P.; Driscoll, T.; Hostetler, J.B.; Nordberg, E.; Shukla, M.; Walenz, B.; Hill, C.A.; Nene, V.M.; et al. A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J. Bacteriol. 2012, 194, 376–394. [Google Scholar] [CrossRef] [Green Version]
- Duron, O.; Binetruy, F.; Noel, V.; Cremaschi, J.; McCoy, K.D.; Arnathau, C.; Plantard, O.; Goolsby, J.; de Leon, A.A.P.; Heylen, D.J.A.; et al. Evolutionary changes in symbiont community structure in ticks. Mol. Ecol. 2017, 26, 2905–2921. [Google Scholar] [CrossRef] [Green Version]
- Al-Khafaji, A.M.; Armstrong, S.D.; Varotto, B.I.; Gaiarsa, S.; Sinha, A.; Li, Z.; Sassera, D.; Carlow, C.K.S.; Epis, S.; Makepeace, B.L. Rickettsia buchneri, symbiont of the deer tick Ixodes scapularis, can colonise the salivary glands of its host. Ticks Tick Borne Dis. 2020, 11, 101299. [Google Scholar] [CrossRef] [PubMed]
- Munderloh, U.G.; Jauron, S.D.; Kurtti, T.J. The tick: A different kind of host for human pathogens. In Tick-Borne Diseases of Humans; Goodman, J.L., Ed.; ASM Press: Washington, DC, USA, 2005; pp. 37–64. [Google Scholar]
- Noda, H.; Munderloh, U.G.; Kurtti, T.J. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl. Environ. Microbiol. 1997, 63, 3926–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelakis, E.; Mediannikov, O.; Jos, S.L.; Berenger, J.M.; Parola, P.; Raoult, D. Candidatus Coxiella massiliensis infection. Emerg. Infect. Dis. 2016, 22, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Eiler, A.; Heinrich, F.; Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012, 6, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.-E.T.; Kim, D.Y.; Sachdeva, R.; Caron, D.A.; Fuhrman, J.A. Top-down controls on bacterial community structure: Microbial network analysis of bacteria, T4-like viruses and protists. ISME J. 2014, 8, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdjeb, L.; Parada, A.; Needham, D.M.; Fuhrman, J.A. Short-term dynamics and interactions of marine protist communities during the spring–summer transition. ISME J. 2018, 12, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Lejal, E.; Moutailler, S.; Šimo, L.; Vayssier-Taussat, M.; Pollet, T. Tick-borne pathogen detection in midgut and salivary glands of adult Ixodes ricinus. Parasites Vectors 2019, 12, 152. [Google Scholar] [CrossRef] [Green Version]
- Buysse, M.; Floriano, A.M.; Gottlieb, Y.; Nardi, T.; Comandatore, F.; Olivieri, E.; Giannetto, A.; Urbina, A.M.P.; Makepeace, B.; Bazzocchi, C.; et al. A dual endosymbiosis drives nutritional adaptation to hematophagy in the invasive tick Hyalomma marginatum. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hunter, D.J.; Torkelson, J.L.; Bodnar, J.; Mortazavi, B.; Laurent, T.; Deason, J.; Thephavongsa, K.; Zhong, J. The Rickettsia endosymbiont of Ixodes pacificus contains all the genes of de novo folate biosynthesis. PLoS ONE 2015, 10, e0144552. [Google Scholar] [CrossRef]
- Steiner, F.E.; Pinger, R.R.; Vann, C.N.; Grindle, N.; Civitello, D.; Clay, K.; Fuqua, C. Infection and co-infection rates of Anaplasma phagocytophilum variants, Babesia spp., Borrelia burgdorferi, and the rickettsial endosymbiont in Ixodes scapularis (Acari: Ixodidae) from sites in Indiana, Maine, Pennsylvania, and Wisconsin. J. Med. Entomol. 2008, 45, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, R.; Tagliafierro, T.; Sameroff, S.; Cucura, D.M.; Oleynik, A.; Che, X.; Jain, K.; Lipkin, W.I. Microbiome analysis of Ixodes scapularis ticks from New York and Connecticut. Ticks Tick Borne Dis. 2019, 10, 894–900. [Google Scholar] [CrossRef]
- Budachetri, K.; Kumar, D.; Crispell, G.; Beck, C.; Dasch, G.; Karim, S. The tick endosymbiont Candidatus Midichloria mitochondrii and selenoproteins are essential for the growth of Rickettsia parkeri in the Gulf Coast tick vector. Microbiome 2018, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Karim, S.; Singh, P.; Ribeiro, J.M. A deep insight into the sialotranscriptome of the gulf coast tick, Amblyomma maculatum. PLoS ONE 2011, 6, e28525. [Google Scholar] [CrossRef]
- Kumar, D.; Raj Sharma, S.; Adegoke, A.; Kenedy, A.; Tuten, H.C.; Li, A.Y.; Karim, S. Recently Evolved Francisella-Like Endosymbiont Outcompetes an Ancient and Evolutionarily Associated Coxiella-Like Endosymbiont in the Lone Star Tick (Amblyomma americanum) Linked to the Alpha-Gal; Research Square: Durham, NC, USA, 2021. [Google Scholar] [CrossRef]
- Adegoke, A.; Kumar, D.; Budachetri, K.; Karim, S. Changes in microbial composition, diversity, and functionality in the Amblyomma maculatum microbiome following infection with Rickettsia parkeri. bioRxiv 2021. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using Microbiome Analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, D.; Downs, L.P.; Adegoke, A.; Machtinger, E.; Oggenfuss, K.; Ostfeld, R.S.; Embers, M.; Karim, S. An Exploratory Study on the Microbiome of Northern and Southern Populations of Ixodes scapularis Ticks Predicts Changes and Unique Bacterial Interactions. Pathogens 2022, 11, 130. https://doi.org/10.3390/pathogens11020130
Kumar D, Downs LP, Adegoke A, Machtinger E, Oggenfuss K, Ostfeld RS, Embers M, Karim S. An Exploratory Study on the Microbiome of Northern and Southern Populations of Ixodes scapularis Ticks Predicts Changes and Unique Bacterial Interactions. Pathogens. 2022; 11(2):130. https://doi.org/10.3390/pathogens11020130
Chicago/Turabian StyleKumar, Deepak, Latoyia P. Downs, Abdulsalam Adegoke, Erika Machtinger, Kelly Oggenfuss, Richard S. Ostfeld, Monica Embers, and Shahid Karim. 2022. "An Exploratory Study on the Microbiome of Northern and Southern Populations of Ixodes scapularis Ticks Predicts Changes and Unique Bacterial Interactions" Pathogens 11, no. 2: 130. https://doi.org/10.3390/pathogens11020130
APA StyleKumar, D., Downs, L. P., Adegoke, A., Machtinger, E., Oggenfuss, K., Ostfeld, R. S., Embers, M., & Karim, S. (2022). An Exploratory Study on the Microbiome of Northern and Southern Populations of Ixodes scapularis Ticks Predicts Changes and Unique Bacterial Interactions. Pathogens, 11(2), 130. https://doi.org/10.3390/pathogens11020130