
From Inflammation to Fibrosis: Novel Insights into the Roles of High Mobility Group Protein Box 1 in Schistosome-Induced Liver Damage

Abstract
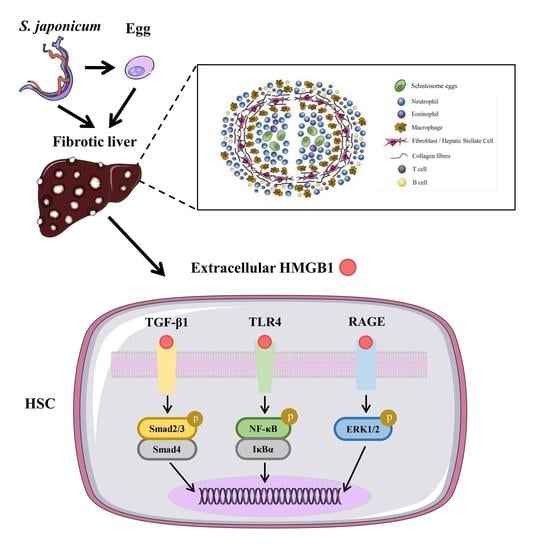
:
{kind=link}
{kind=link}
{kind=link}
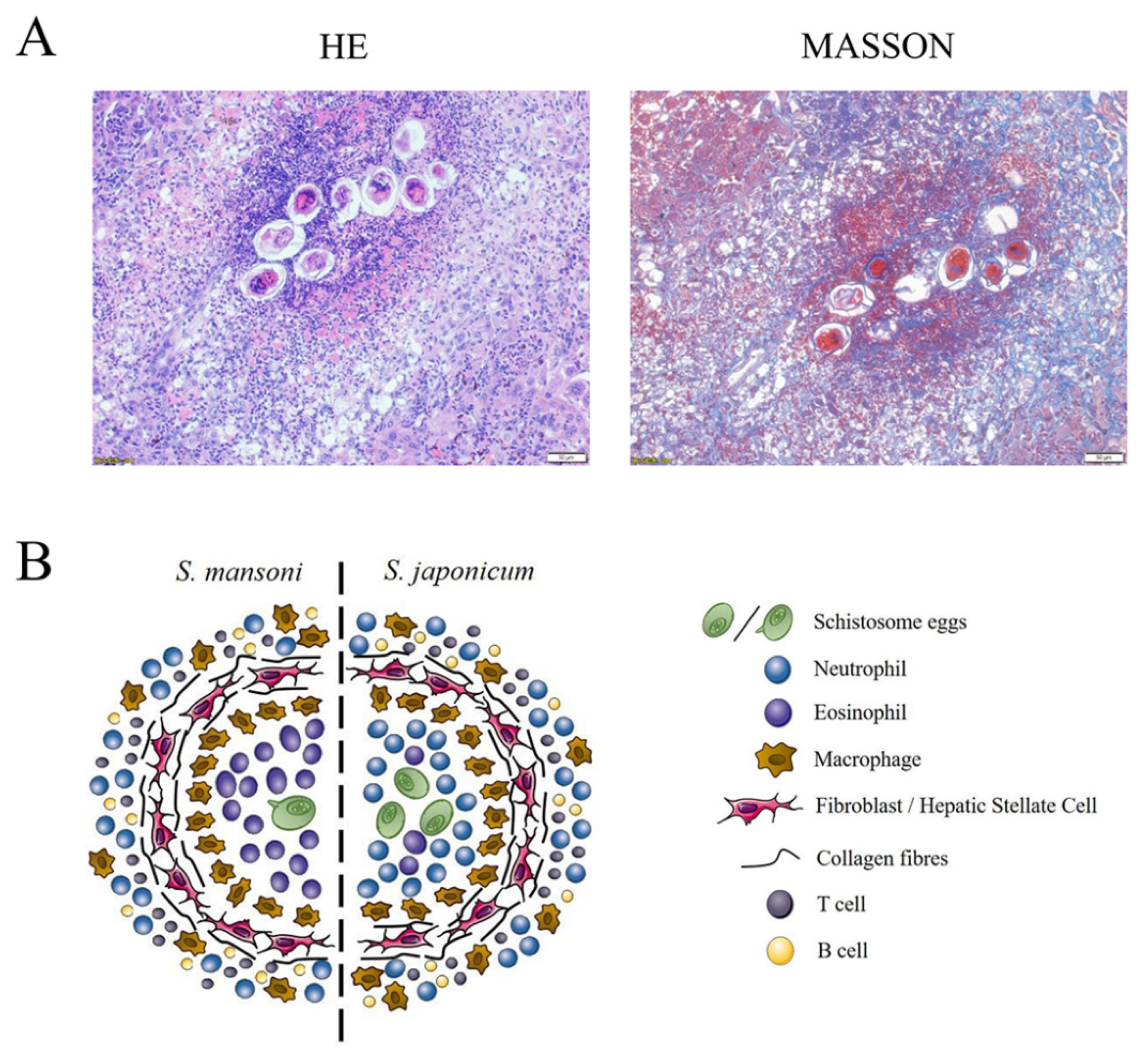{kind=link}
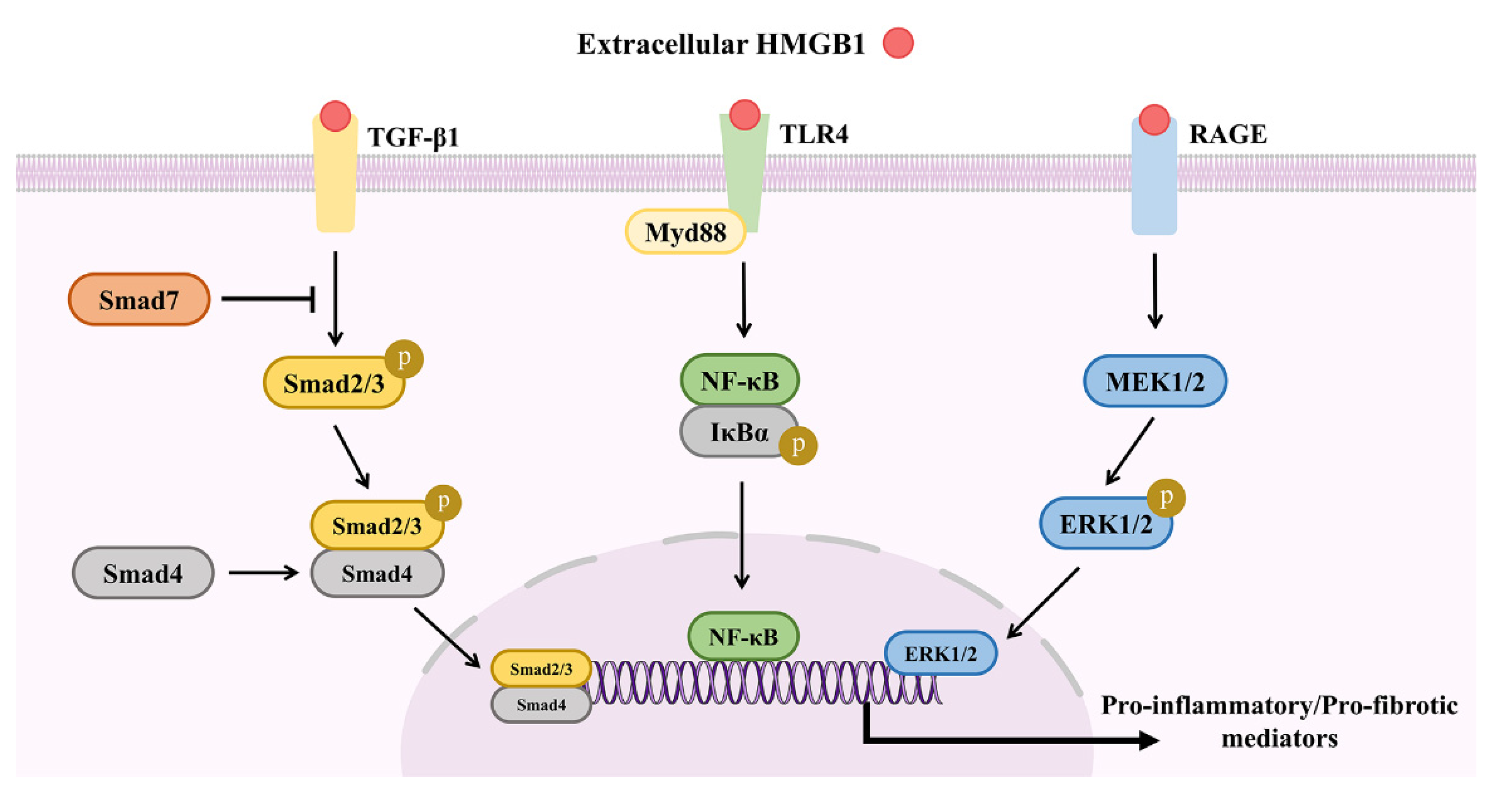{kind=link}
1. Introduction
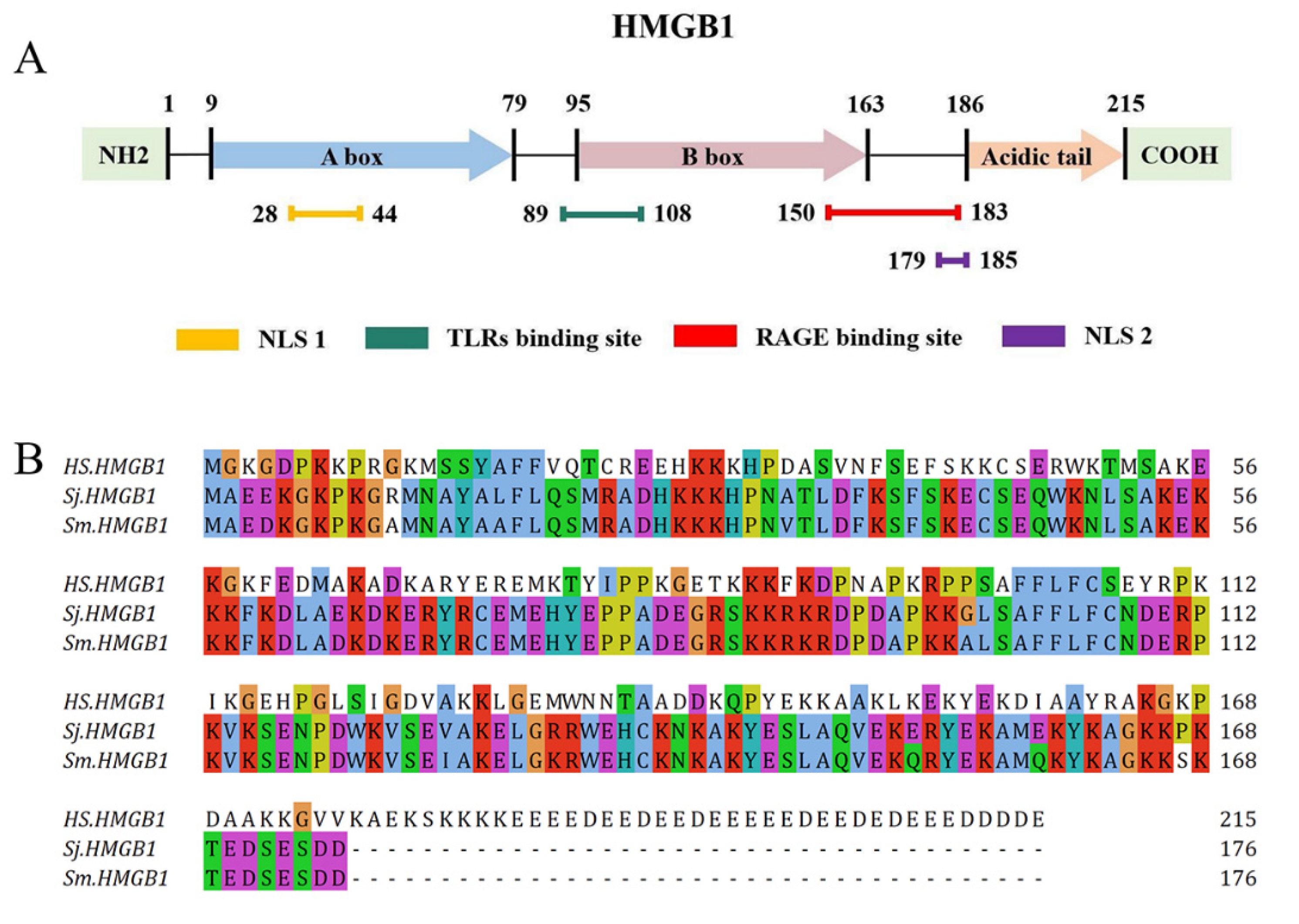
2. HMGB1 Structure and Receptors
2.1. HMGB1 Structure
2.2. HMGB1 Receptors
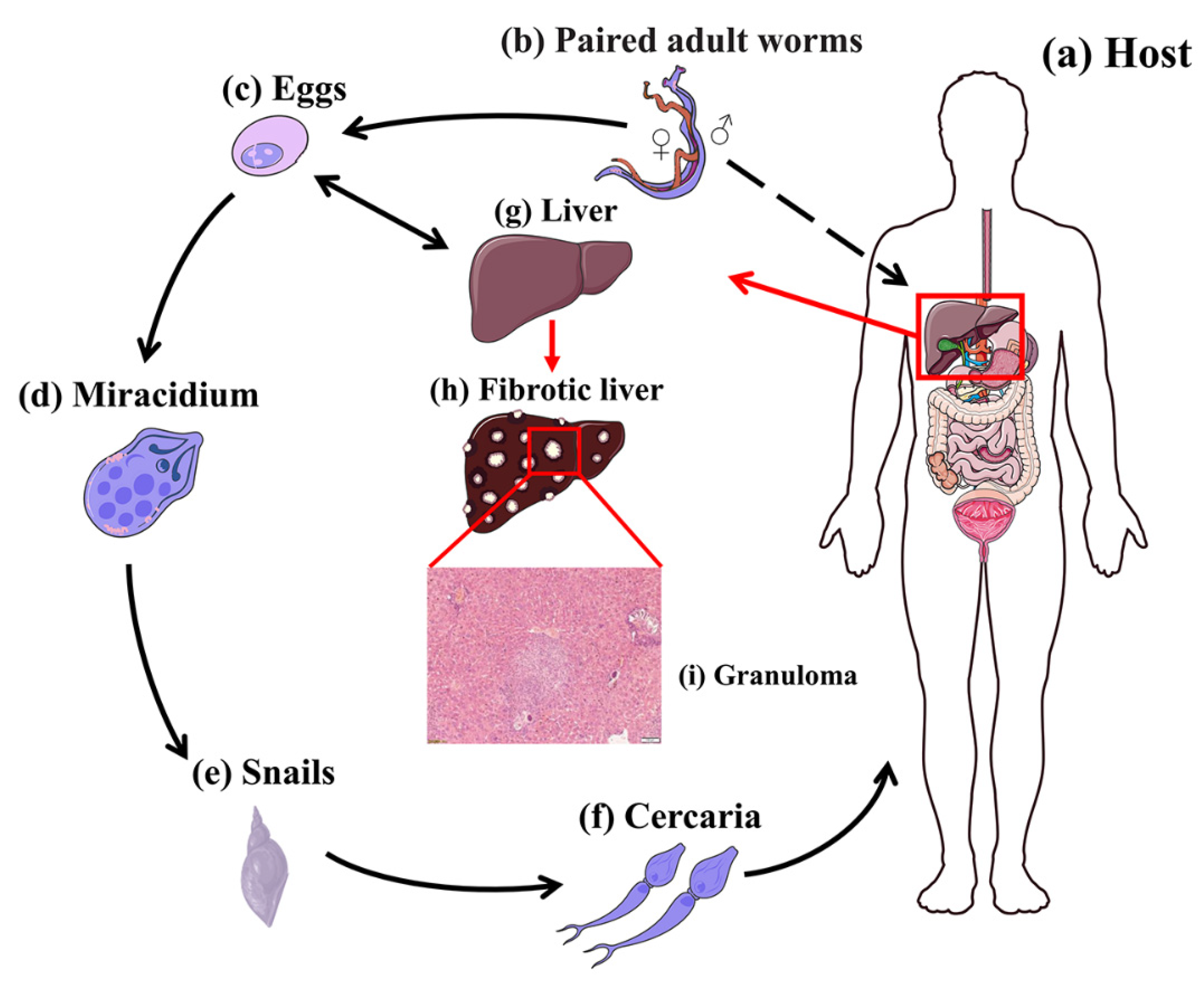
3. Schistosome-Induced Liver Damage
3.1. Life Cycles of Schistosomes
3.2. Formation of Liver Granulomas
4. HMGB1 and Schistosome-Induced Liver Damage
4.1. HMGB1 and Liver Inflammation
4.2. HMGB1 and Liver Fibrosis
5. HMGB1 as a Therapeutic Target
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO. TDR Strategic Direction for Research: Schistosomiasis; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Boissier, J.; Grech-Angelini, S.; Webster, B.L.; Allienne, J.F.; Huyse, T.; Mas-Coma, S.; Toulza, E.; Barré-Cardi, H.; Rollinson, D.; Kincaid-Smith, J.; et al. Outbreak of urogenital schistosomiasis in Corsica (France): An epidemiological case study. Lancet 2016, 16, 971–979. [Google Scholar] [CrossRef]
- Chitsulo, L.; Loverde, P.; Engels, D. Focus: Schistosomiasis. Nat. Rev. Microbiol. 2004, 2, 12. [Google Scholar] [CrossRef]
- Nation, C.S.; Da’dara, A.A.; Marchant, J.K.; Skelly, P.J. Schistosome migration in the definitive host. PLoS Negl. Trop. Dis. 2020, 14, e0007951. [Google Scholar] [CrossRef]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Cheever, A.W.; Macedonia, J.G.; Mosimann, J.E.; Cheever, E.A. Kinetics of egg production and egg excretion by Schistosoma mansoni and S. japonicum in mice infected with a single pair of worms. Am. J. Trop. Med. Hyg. 1994, 50, 281–295. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Tang, R.; Sun, Y.; Wang, Y.G.; Zhen, K.Y.; Zhang, D.M.; Pan, W.Q. MicroR-146 blocks the activation of M1 macrophage by targeting signal transducer and activator of transcription 1 in hepatic schistosomiasis. EBioMedicine 2016, 13, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Chuah, C.; Jones, M.K.; Burke, M.L.; McManus, D.P.; Gobert, G.N. Cellular and chemokine-mediated regulation in schistosome-induced hepatic pathology. Trends Parasitol. 2014, 30, 141–150. [Google Scholar] [CrossRef]
- Soares Magalhães, R.J.; Barnett, A.G.; Clements, A.C. Geographical analysis of the role of water supply and sanitation in the risk of helminth infections of children in West Africa. Proc. Natl. Acad. Sci. USA 2011, 108, 20084–20089. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.D.; Chen, H.G.; Guo, J.G.; Zeng, X.J.; Hong, X.L.; Xiong, J.J.; Wu, X.H.; Wang, X.H.; Wang, L.Y.; Xia, G.; et al. A strategy to control transmission of Schistosoma japonicum in China. N. Engl. J. Med. 2009, 360, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Fenwick, A.; Savioli, L.; Engels, D.; Robert Bergquist, N.; Todd, M.H. Drugs for the control of parasitic diseases: Current status and development in schistosomiasis. Trends Parasitol. 2003, 19, 509–515. [Google Scholar] [CrossRef]
- Hagen, J.; Scheerlinck, J.P.; Gasser, R.B. Knocking down schistosomes—Promise for lentiviral transduction in parasites. Trends Parasitol. 2015, 31, 324–332. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Wei, Y.; Liu, H.; Zhang, J.; Shen, Y.; Cao, J. Functional Inhibition of Natural Killer Cells in a BALB/c Mouse Model of Liver Fibrosis Induced by Schistosoma japonicum Infection. Front. Cell. Infect. Microbiol. 2020, 10, 598987. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Vicentino, A.R.R.; Carneiro, V.C.; Allonso, D.; Guilherme, R.F.; Benjamim, C.F.; Dos Santos, H.A.M.; Xavier, F.; Pyrrho, A.D.S.; Gomes, J.A.S.; Fonseca, M.C.; et al. Emerging Role of HMGB1 in the Pathogenesis of Schistosomiasis Liver Fibrosis. Front. Immunol. 2018, 9, 1979. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, G.; Zhang, J.; Zheng, T.; Chen, Q.; Zhang, Y.; Yang, F.; Wang, C.; Nie, H.; Zheng, B.; et al. Sodium butyrate ameliorates Schistosoma japonicum-induced liver fibrosis by inhibiting HMGB1 expression. Exp. Parasitol. 2021, 231, 108171. [Google Scholar] [CrossRef]
- Ge, W.-S. Inhibition of high-mobility group box 1 expression by siRNA in rat hepatic stellate cells. World J. Gastroenterol. 2011, 17, 4090. [Google Scholar] [CrossRef]
- Huang, H.; Nace, G.W.; McDonald, K.A.; Tai, S.; Klune, J.R.; Rosborough, B.R.; Ding, Q.; Loughran, P.; Zhu, X.; Beer-Stolz, D.; et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014, 59, 1984–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, F.M.; de Abreu da Silva, I.C.; Rumjanek, F.D.; Dias-Neto, E.; Guimarães, P.E.; Verjovski-Almeida, S.; Stros, M.; Fantappié, M.R. Cloning the genes and DNA binding properties of High Mobility Group B1 (HMGB1) proteins from the human blood flukes Schistosoma mansoni and Schistosoma japonicum. Gene 2006, 377, 33–45. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J. Biol. Chem. 2002, 277, 38635–38646. [Google Scholar] [CrossRef] [Green Version]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [CrossRef]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nε-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, K.; Tsuneyama, K.; Abdel Aziz, H.O.; Takahashi, H.; Murai, Y.; Cui, Z.G.; Fujimoto, M.; Kato, I.; Hiraga, K.; Hsu, D.K.; et al. Disrupted galectin-3 causes non-alcoholic fatty liver disease in male mice. J. Pathol. 2006, 210, 469–477. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, X.; Su, X.; Liu, X.; Ren, K.; Ning, C.; Zhang, Q.; Zhang, S. Daphnes Cortex and its licorice-processed products suppress inflammation via the TLR4/NF-κB/NLRP3 signaling pathway and regulation of the metabolic profile in the treatment of rheumatoid arthritis. J. Ethnopharmacol. 2022, 283, 114657. [Google Scholar] [CrossRef]
- Cai, J.; Li, J.; Zhou, Y.; Wang, J.; Li, J.; Cui, L.; Meng, X.; Zhu, G.; Wang, H. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J. Cell. Mol. Med. 2020, 24, 3460–3468. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Ding, T.; Fang, L.; Cui, L.; Li, J.; Meng, X.; Zhu, G.; Qian, C.; Wang, H.; Li, J. Organic Selenium Ameliorates Staphylococcus aureus-Induced Mastitis in Rats by Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. Front. Vet. Sci. 2020, 7, 443. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Yang, J.; Wang, L.; Tan, X.; Hu, X.; Sun, L.; Chen, S.; Zhu, L.; Chen, X.; et al. Liraglutide protects against lethal renal ischemia-reperfusion injury by inhibiting high-mobility group box 1 nuclear-cytoplasmic translocation and release. Pharmacol. Res. 2021, 173, 105867. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Jakaria, M.; Kim, I.S.; Kim, J.; Haque, M.E.; Choi, D.K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qu, H.; Zhang, H.; Zhong, X. Prunella vulgaris L. Attenuates Experimental Autoimmune Thyroiditis by Inhibiting HMGB1/TLR9 Signaling. Drug Des. Dev. Ther. 2021, 15, 4559–4574. [Google Scholar] [CrossRef] [PubMed]
- Stensgaard, A.S.; Vounatsou, P.; Sengupta, M.E.; Utzinger, J. Schistosomes, snails and climate change: Current trends and future expectations. Acta Trop. 2019, 190, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Hailegebriel, T.; Nibret, E.; Munshea, A. Prevalence of Schistosoma mansoni and S. haematobium in Snail Intermediate Hosts in Africa: A Systematic Review and Meta-analysis. J. Trop. Med. 2020, 2020, 8850840. [Google Scholar] [CrossRef]
- He, Y.X.; Chen, L.; Ramaswamy, K. Schistosoma mansoni, S. haematobium, and S. japonicum: Early events associated with penetration and migration of schistosomula through human skin. Exp. Parasitol. 2002, 102, 99–108. [Google Scholar] [CrossRef]
- He, Y.X.; Salafsky, B.; Ramaswamy, K. Comparison of skin invasion among three major species of Schistosoma. Trends Parasitol. 2005, 21, 201–203. [Google Scholar] [CrossRef]
- Miller, P.; Wilson, R.A. Migration of the schistosomula of Schistosoma mansoni from the lungs to the hepatic portal system. Parasitology 1980, 80, 267–288. [Google Scholar] [CrossRef]
- Wheater, P.R.; Wilson, R.A. Schistosoma mansoni: A histological study of migration in the laboratory mouse. Parasitology 1979, 79, 49–62. [Google Scholar] [CrossRef]
- Ito, J. Studies on the host-parasite relationships of Schistosoma japonicum in common laboratory animals. Jpn. J. Med. Sci. Biol. 1955, 8, 43–62. [Google Scholar] [CrossRef] [Green Version]
- Georgi, J.R.; Wade, S.E.; Dean, D.A. Attrition and temporal distribution of Schistosoma mansoni and S. haematobium schistosomula in laboratory mice. Parasitology 1986, 93 Pt 1, 55–70. [Google Scholar] [CrossRef] [PubMed]
- van Oordt, B.E.; Tielens, A.G.; van den Bergh, S.G. The energy metabolism of Schistosoma mansoni during its development in the hamster. Parasitol. Res. 1988, 75, 31–35. [Google Scholar] [CrossRef]
- Cheever, A.W. A quantitative post-mortem study of Schistosomiasis mansoni in man. Am. J. Trop. Med. Hyg. 1968, 17, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.G. Relative distribution of Schistosoma japonicum eggs in the intestine of man: A subject of inconsistency. Acta Trop. 1991, 48, 163–171. [Google Scholar] [CrossRef]
- Gelfand, M.; Ross, W.F. I. The distribution of schistosome ova in the alimentary tract in subjects of bilharziasis. Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 215–217. [Google Scholar] [CrossRef]
- Alves, W. The distribution of Schistosoma eggs in human tissues. Bull. World Health Organ. 1958, 18, 1092–1097. [Google Scholar] [PubMed]
- Lee, K.F.; Hsueh, S.; Tang, M.H. Schistosomiasis of the ovary with endometriosis and corpus hemorrhagicum: A case report. Chang. Gung Med. J. 2000, 23, 438–441. [Google Scholar] [PubMed]
- Carpenter, C.B.; Mozley, P.D.; Lewis, N.G. Schistosomiasis japonica involvement of the female genital tract. Jama 1964, 188, 647–650. [Google Scholar] [CrossRef]
- Yu, Z.; Wei, C.; Wang, Y.; Ye, Z.; Wang, Z.; Chen, Z.; Ni, L.; Yang, S.; Gui, Y.; Guan, Z.; et al. Prostatic Schistosoma japonicum with atypical immunophenotyping of individual glandular tubes: A case report and review of the literature. Southeast Asian J. Trop. Med. Public Health 2013, 44, 568–573. [Google Scholar]
- Wen, S.C.H.; Anderson, R.; Ryan, M.M.; Kumbla, S.; Wray, A.; Steer, A. Pediatric Neuroschistosomiasis: A Case Report and Review of the Literature. J. Pediatric Infect. Dis. Soc. 2019, 8, 489–491. [Google Scholar] [CrossRef]
- Dastoli, P.A.; Leite, A.L.; da Costa, M.D.S.; Nicácio, J.M.; Pinho, R.S.; Ferrarini, M.A.G.; Cavalheiro, S. Medullary neuroschistosomiasis in adolescence: Case report and literature review. Child’s Nerv. Syst. 2021, 37, 2735–2741. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yu, C.X.; Song, L.J.; Yin, X.R.; Wang, J.; Jin, Y.; Shuan, S.; Zhang, W.; Gao, H.; Xu, Y.L.; et al. Cloning and function analysis of high mobility group box 1 (HMGB1) protein of Schistosoma japonicum (Mainland strain). Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 2014, 26, 153–159. [Google Scholar] [PubMed]
- Gelfand, M.; Ross, W.F. II. The distribution of schistosome ova in the genito-urinary tract in subjects of bilharziasis. Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 218–220. [Google Scholar] [CrossRef]
- Gelfand, M.; Ross, M.D.; Blair, D.M.; Weber, M.C. Distribution and extent of schistosomiasis in female pelvic organs, with special reference to the genital tract, as determined at autopsy. Am. J. Trop. Med. Hyg. 1971, 20, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, M.; Ross, C.M.; Blair, D.M.; Castle, W.M.; Weber, M.C. Schistosomiasis of the male pelvic organs. Severity of infection as determined by digestion of tissue and histologic methods in 300 cadavers. Am. J. Trop. Med. Hyg. 1970, 19, 779–784. [Google Scholar] [CrossRef]
- Andrade, Z.A. Schistosomiasis and liver fibrosis. Parasite Immunol. 2009, 31, 656–663. [Google Scholar] [CrossRef]
- Ashton, P.D.; Harrop, R.; Shah, B.; Wilson, R.A. The schistosome egg: Development and secretions. Parasitology 2001, 122, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Fabre, V.; Wu, H.; PondTor, S.; Coutinho, H.; Acosta, L.; Jiz, M.; Olveda, R.; Cheng, L.; White, E.S.; Jarilla, B.; et al. Tissue inhibitor of matrix-metalloprotease-1 predicts risk of hepatic fibrosis in human Schistosoma japonicum infection. J. Infect. Dis. 2011, 203, 707–714. [Google Scholar] [CrossRef]
- Coutinho, H.M.; Acosta, L.P.; Wu, H.W.; McGarvey, S.T.; Su, L.; Langdon, G.C.; Jiz, M.A.; Jarilla, B.; Olveda, R.M.; Friedman, J.F.; et al. Th2 cytokines are associated with persistent hepatic fibrosis in human Schistosoma japonicum infection. J. Infect. Dis. 2007, 195, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Carson, J.P.; Ramm, G.A.; Robinson, M.W.; McManus, D.P.; Gobert, G.N. Schistosome-Induced Fibrotic Disease: The Role of Hepatic Stellate Cells. Trends Parasitol. 2018, 34, 524–540. [Google Scholar] [CrossRef]
- Wilson, M.S.; Mentink-Kane, M.M.; Pesce, J.T.; Ramalingam, T.R.; Thompson, R.; Wynn, T.A. Immunopathology of schistosomiasis. Immunol. Cell Biol. 2007, 85, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Brunet, L.R.; Finkelman, F.D.; Cheever, A.W.; Kopf, M.A.; Pearce, E.J. IL-4 protects against TNF-alpha-mediated cachexia and death during acute schistosomiasis. J. Immunol. 1997, 159, 777–785. [Google Scholar] [PubMed]
- Warren, K.S.; Domingo, E.O. Granuloma formation around Schistosoma mansoni, S. haematobium, and S. japonicum eggs. Size and rate of development, cellular composition, cross-sensitivity, and rate of egg destruction. Am. J. Trop. Med. Hyg. 1970, 19, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Schramm, G.; Gronow, A.; Knobloch, J.; Wippersteg, V.; Grevelding, C.G.; Galle, J.; Fuller, H.; Stanley, R.G.; Chiodini, P.L.; Haas, H.; et al. IPSE/alpha-1: A major immunogenic component secreted from Schistosoma mansoni eggs. Mol. Biochem. Parasitol. 2006, 147, 9–19. [Google Scholar] [CrossRef]
- Schramm, G.; Falcone, F.H.; Gronow, A.; Haisch, K.; Mamat, U.; Doenhoff, M.J.; Oliveira, G.; Galle, J.; Dahinden, C.A.; Haas, H. Molecular characterization of an interleukin-4-inducing factor from Schistosoma mansoni eggs. J. Biol. Chem. 2003, 278, 18384–18392. [Google Scholar] [CrossRef] [Green Version]
- Chuah, C.; Jones, M.K.; Burke, M.L.; Owen, H.C.; Anthony, B.J.; McManus, D.P.; Ramm, G.A.; Gobert, G.N. Spatial and temporal transcriptomics of Schistosoma japonicum-induced hepatic granuloma formation reveals novel roles for neutrophils. J. Leukoc. Biol. 2013, 94, 353–365. [Google Scholar] [CrossRef]
- Washington, K.; Wright, K.; Shyr, Y.; Hunter, E.B.; Olson, S.; Raiford, D.S. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum. Pathol. 2000, 31, 822–828. [Google Scholar] [CrossRef]
- Vera, M.; Nieto, N. Hepatic stellate cells and alcoholic liver disease. Rev. Esp. Enferm. Dig. 2006, 98, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Bartley, P.B.; Ramm, G.A.; Jones, M.K.; Ruddell, R.G.; Li, Y.; McManus, D.P. A contributory role for activated hepatic stellate cells in the dynamics of Schistosoma japonicum egg-induced fibrosis. Int. J. Parasitol. 2006, 36, 993–1001. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Alisi, A.; He, X.; Pu, G.; Tang, R.; Zhang, D.; Pan, W. Activation of Nuclear Factor Kappa B in the Hepatic Stellate Cells of Mice with Schistosomiasis Japonica. PLoS ONE 2014, 9, e104323. [Google Scholar] [CrossRef] [Green Version]
- Ragheb, S.; Boros, D.L. Characterization of granuloma T lymphocyte function from Schistosoma mansoni-infected mice. J. Immunol. 1989, 142, 3239–3246. [Google Scholar] [PubMed]
- Song, E.; Ouyang, N.; Hörbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell. Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Lian, Y.J.; Gong, H.; Wu, T.Y.; Su, W.J.; Zhang, Y.; Yang, Y.Y.; Peng, W.; Zhang, T.; Zhou, J.R.; Jiang, C.L.; et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain Behav. Immun. 2017, 59, 322–332. [Google Scholar] [CrossRef]
- Yu, R.; Jiang, S.; Tao, Y.; Li, P.; Yin, J.; Zhou, Q. Inhibition of HMGB1 improves necrotizing enterocolitis by inhibiting NLRP3 via TLR4 and NF-kappaB signaling pathways. J. Cell. Physiol. 2019, 234, 13431–13438. [Google Scholar] [CrossRef]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [Green Version]
- Sepehri, Z.; Kiani, Z.; Kohan, F.; Alavian, S.M.; Ghavami, S. Toll like receptor 4 and hepatocellular carcinoma; A systematic review. Life Sci. 2017, 179, 80–87. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Sang, R.; Yu, Y.; Ge, B.; Xu, L.; Wang, Z.; Zhang, X. Taraxasterol from Taraxacum prevents concanavalin A-induced acute hepatic injury in mice via modulating TLRs/NF-κB and Bax/Bc1-2 signalling pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3929–3937. [Google Scholar] [CrossRef] [Green Version]
- Nabih, E.S.; El-Kharashi, O.A. Targeting HMGB1/TLR4 axis and miR-21 by rosuvastatin: Role in alleviating cholestatic liver injury in a rat model of bile duct ligation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 37–43. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Liu, Q.; Zhang, W.; Wan, Y.; Huang, C.; Zhu, X. Ursolic acid reverses liver fibrosis by inhibiting NOX4/NLRP3 inflammasome pathways and bacterial dysbiosis. Gut Microbes 2021, 13, 1972746. [Google Scholar] [CrossRef]
- Wang, L.; Liao, Y.; Yang, R.; Yu, Z.; Zhang, L.; Zhu, Z.; Wu, X.; Shen, J.; Liu, J.; Xu, L.; et al. Sja-miR-71a in Schistosome egg-derived extracellular vesicles suppresses liver fibrosis caused by schistosomiasis via targeting semaphorin 4D. J. Extracell. Vesicles 2020, 9, 1785738. [Google Scholar] [CrossRef]
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Yin, X.M. Role of High-Mobility Group Box-1 in Liver Pathogenesis. Int. J. Mol. Sci. 2019, 20, 5314. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Gao, J.; Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 2014, 18, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Lin, Y.C.; Tsai, M.S.; Sun, C.K.; Yuan, S.S.; Chang, C.Y.; Jawan, B.; Lee, P.H. Involvement of the nuclear high mobility group B1 peptides released from injured hepatocytes in murine hepatic fibrogenesis. Biochim. Biophys. Acta 2014, 1842, 1720–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albayrak, A.; Uyanik, M.H.; Cerrah, S.; Altas, S.; Dursun, H.; Demir, M.; Uslu, H. Is HMGB1 a new indirect marker for revealing fibrosis in chronic hepatitis and a new therapeutic target in treatment? Viral Immunol. 2010, 23, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.R.; Yang, C.Q.; Shi, Q. Transforming growth factor-β1 induced epithelial-mesenchymal transition in hepatic fibrosis. Hepato-Gastroenterology 2012, 59, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Kweon, Y.O.; Frederick, J.P.; Wang, X.F.; Rippe, R.A.; Brenner, D.A. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 2001, 34, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Zhang, Y. MiR-92d-3p suppresses the progression of diabetic nephropathy renal fibrosis by inhibiting the C3/HMGB1/TGF-β1 pathway. Biosci. Rep. 2021, 41, BSR20203131. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, F.; Zhang, A. Arsenic-induced lung inflammation and fibrosis in a rat model: Contribution of the HMGB1/RAGE, PI3K/AKT, and TGF-β1/SMAD pathways. Toxicol. Appl. Pharmacol. 2021, 432, 115757. [Google Scholar] [CrossRef]
- Arriazu, E.; Ge, X.; Leung, T.M.; Magdaleno, F.; Lopategi, A.; Lu, Y.; Kitamura, N.; Urtasun, R.; Theise, N.; Antoine, D.J.; et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017, 66, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Dela Cruz, R.; Theise, N.; Nieto, N. High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyganowska-Swiatkowska, M.; Nohawica, M.; Grocholewicz, K.; Nowak, G. Influence of Herbal Medicines on HMGB1 Release, SARS-CoV-2 Viral Attachment, Acute Respiratory Failure, and Sepsis. A Literature Review. Int. J. Mol. Sci. 2020, 21, 4639. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Chen, X.; Ling, Y.; Wei, Y.; Tang, J.; Ren, Y.; Zhang, B.; Jiang, F.; Li, H.; Wang, R.; Wen, W.; et al. Dual regulation of HMGB1 by combined JNK1/2-ATF2 axis with miR-200 family in nonalcoholic steatohepatitis in mice. FASEB J. 2018, 32, 2722–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entezari, M.; Weiss, D.J.; Sitapara, R.; Whittaker, L.; Wargo, M.J.; Li, J.; Wang, H.; Yang, H.; Sharma, L.; Phan, B.D.; et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas Aeruginosa pneumonia in cystic fibrosis. Mol. Med. 2012, 18, 477–485. [Google Scholar] [CrossRef]
- Iino, S.; Tango, T.; Matsushima, T.; Toda, G.; Miyake, K.; Hino, K.; Kumada, H.; Yasuda, K.; Kuroki, T.; Hirayama, C.; et al. Therapeutic effects of stronger neo-minophagen C at different doses on chronic hepatitis and liver cirrhosis. Hepatol. Res. 2001, 19, 31–40. [Google Scholar] [CrossRef]
- Ogiku, M.; Kono, H.; Hara, M.; Tsuchiya, M.; Fujii, H. Glycyrrhizin prevents liver injury by inhibition of high-mobility group box 1 production by Kupffer cells after ischemia-reperfusion in rats. J. Pharmacol. Exp. Ther. 2011, 339, 93–98. [Google Scholar] [CrossRef]
- Gwak, G.Y.; Moon, T.G.; Lee, D.H.; Yoo, B.C. Glycyrrhizin attenuates HMGB1-induced hepatocyte apoptosis by inhibiting the p38-dependent mitochondrial pathway. World J. Gastroenterol. 2012, 18, 679–684. [Google Scholar] [CrossRef]
- Gowda, P.; Patrick, S.; Joshi, S.D.; Kumawat, R.K.; Sen, E. Glycyrrhizin prevents SARS-CoV-2 S1 and Orf3a induced high mobility group box 1 (HMGB1) release and inhibits viral replication. Cytokine 2021, 142, 155496. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, H.; Gui, X.; Hou, L.; Lv, R.; Jin, Y. From Inflammation to Fibrosis: Novel Insights into the Roles of High Mobility Group Protein Box 1 in Schistosome-Induced Liver Damage. Pathogens 2022, 11, 289. https://doi.org/10.3390/pathogens11030289
Zhong H, Gui X, Hou L, Lv R, Jin Y. From Inflammation to Fibrosis: Novel Insights into the Roles of High Mobility Group Protein Box 1 in Schistosome-Induced Liver Damage. Pathogens. 2022; 11(3):289. https://doi.org/10.3390/pathogens11030289
Chicago/Turabian StyleZhong, Haoran, Xiang Gui, Ling Hou, Rongxue Lv, and Yamei Jin. 2022. "From Inflammation to Fibrosis: Novel Insights into the Roles of High Mobility Group Protein Box 1 in Schistosome-Induced Liver Damage" Pathogens 11, no. 3: 289. https://doi.org/10.3390/pathogens11030289
APA StyleZhong, H., Gui, X., Hou, L., Lv, R., & Jin, Y. (2022). From Inflammation to Fibrosis: Novel Insights into the Roles of High Mobility Group Protein Box 1 in Schistosome-Induced Liver Damage. Pathogens, 11(3), 289. https://doi.org/10.3390/pathogens11030289