
A Tale of Two Fimbriae: How Invasion of Dendritic Cells by Porphyromonas gingivalis Disrupts DC Maturation and Depolarizes the T-Cell-Mediated Immune Response


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Periodontitis
1.2. P. gingivalis Fimbriae and the Prevalence of Periodontal Disease
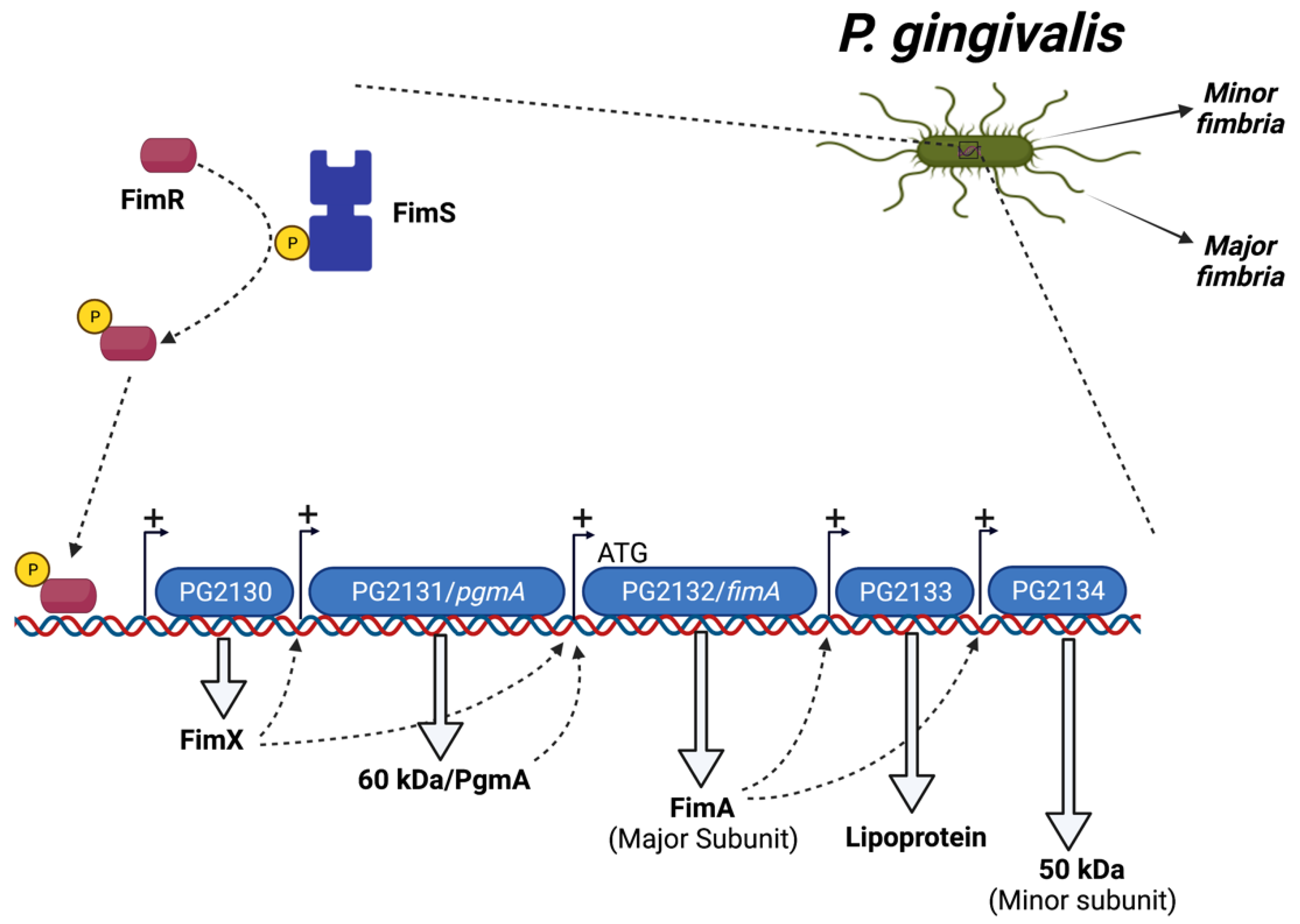
1.3. Regulation of P. gingivalis Fimbriae
1.4. Dendritic Cells: Revisiting the Dogma of DC Subsets and Differentiation
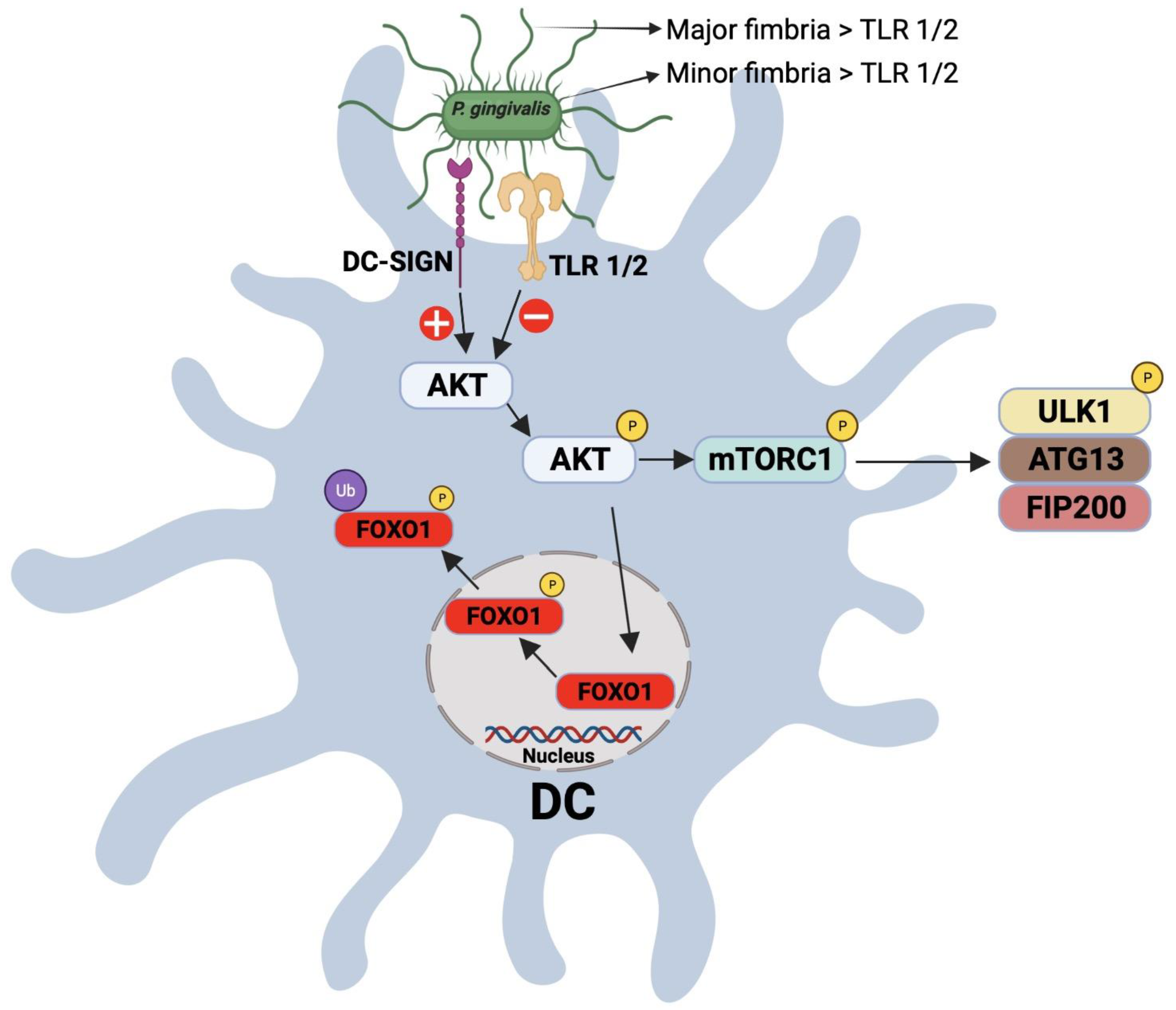
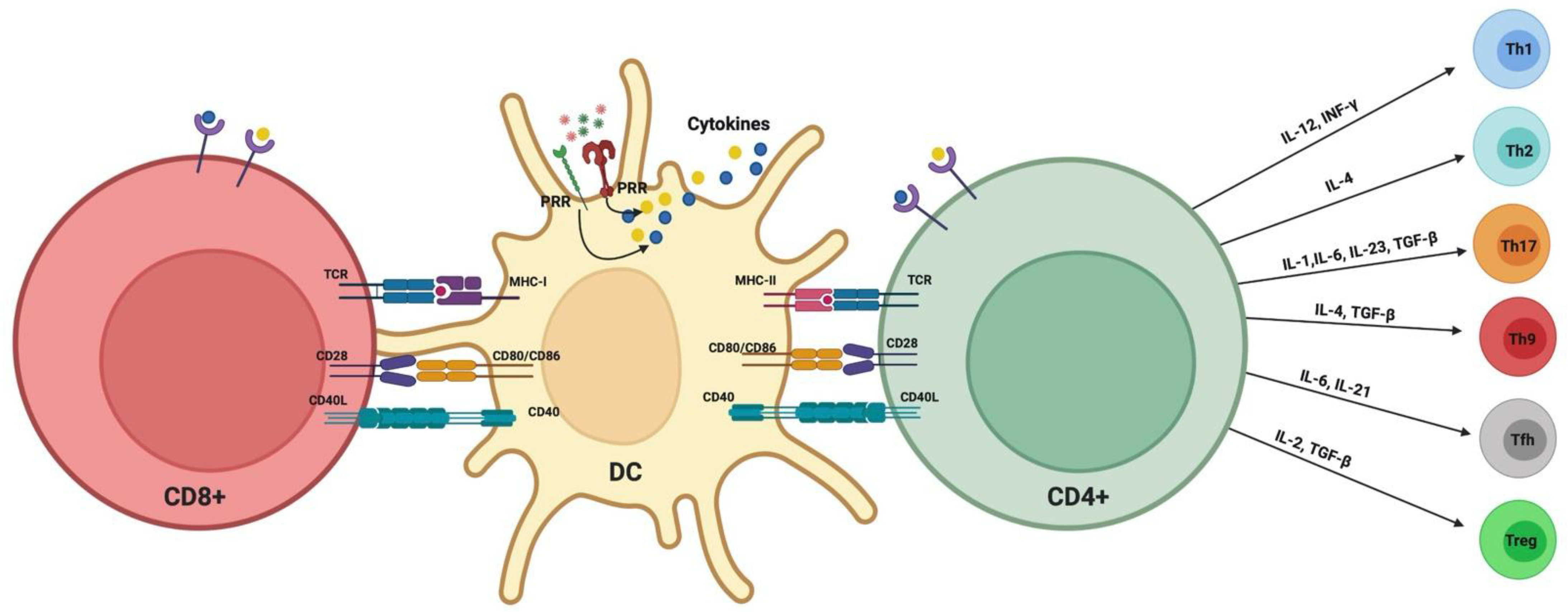
1.5. Modulation of DC–T Cell Interaction by P. gingivalis
1.6. Significance of DC–T Cells’ Clusters in Periodontitis: Oral Lymphoid Foci
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ebersole, J.L.; Dawson, D.A., III; Huja, P.E.; Pandruvada, S.; Basu, A.; Nguyen, L.; Zhang, Y.; Gonzalez, O.A. Age and Periodontal Health—Immunological View. Curr. Oral. Health Rep. 2018, 5, 229–241. [Google Scholar] [CrossRef]
- Elsayad, R.; Elashiry, M.; Lui, Y.; El-Awady, A.; Hamrick, M.; Cutler, C.W. Porphyromonas gingivalis provokes exosome secretion and paracrine immune senescence in bystander dendritic cells. Front. Cell. Infect. Microbiol. 2021, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015, 86, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagaitkar, J.; Daep, C.A.; Patel, C.K.; Renaud, D.E.; De Muth, N.R.; Scott, D.A. Tobacco Smoke Augments Porphyromonas gingivalis—Streptococcus gordonii Biofilm Formation. PLoS ONE 2011, 6, e27386. [Google Scholar] [CrossRef] [PubMed]
- Brook, I. The Impact of Smoking on Oral and Nasopharyngeal Bacterial Flora. J. Dent. Res. 2011, 90, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Haffajee, A.D.; Socransky, S.S. Relationship of cigarette smoking to the subgingival microbiota. J. Clin. Periodontol. 2001, 28, 377–388. [Google Scholar] [CrossRef]
- Kinane, D.F.; Chestnutt, I.G. Smoking and periodontal disease. Crit. Rev. Oral. Biol. Med. 2000, 11, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, M.; Tanno-Nakanishi, M.; Yamada, S.; Okuda, K.; Ishihara, K. Effect of smoking on subgingival microflora of patients with periodontitis in Japan. BMC Oral Health 2011, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.S.; Matthews, C.R.; Joshi, V.; de Jager, M.; Aspiras, M. Tobacco Smoking Affects Bacterial Acquisition and Colonization in Oral Biofilms. Infect. Immun. 2011, 79, 4730–4738. [Google Scholar] [CrossRef] [Green Version]
- Palmer, R.M.; Wilson, R.F.; Hasan, A.S.; Scott, D.A. Mechanisms of action of environmental factors—tobacco smoking. J. Clin. Periodontol. 2005, 32, 180–195. [Google Scholar] [CrossRef]
- Shchipkova, A.; Nagaraja, H.; Kumar, P. Subgingival Microbial Profiles of Smokers with Periodontitis. J. Dent. Res. 2010, 89, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.D.; Koch, G.G.; Offenbacher, S. Incidence of attachment loss over 3 years in older adults—new and progressing lesions. Community Dent. Oral Epidemiol. 1995, 23, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.D.; Cusmano, L.; Green-Helms, W.; Koch, G.G.; Offenbacher, S. A 5-year study of attachment loss in community-dwelling older adults: Incidence density. J. Periodontal Res. 1997, 32, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Machtei, E.E.; Hausmann, E.; Dunford, R.; Grossi, S.; Ho, A.; Davis, G.; Chandler, J.; Zambon, J.; Genco, R.J. Longitudinal study of predictive factors for periodontal disease and tooth loss. J. Clin. Periodontol. 1999, 26, 374–380. [Google Scholar] [CrossRef]
- Norderyd, O.; Hugoson, A.; Grusovin, G. Risk of severe periodontal disease in a Swedish adult population. J. Clin. Periodontol. 1999, 26, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wolff, L.; Aeppli, D.; Guo, Z.; Luan, W.-M.; Baelum, V.; Fejeskov, O. Cigarette smoking, salivary/gingival crevicular fluid cotinine and periodontal status A 10-year longitudinal study. J. Clin. Periodontol. 2001, 28, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Yoshihara, A.; Hirotomi, T.; Ando, Y.; Miyazaki, H. Risk factors for periodontal disease progression among elderly people. J. Clin. Periodontol. 2002, 29, 592–597. [Google Scholar] [CrossRef]
- Paulander, J.; Wennström, J.L.; Axelsson, P.; Lindhe, J. Some risk factors for periodontal bone loss in 50-year-old individuals. A 10-year cohort study. J. Clin. Periodontol. 2004, 31, 489–496. [Google Scholar] [CrossRef]
- Ah, M.K.B.; Johnson, G.K.; Kaldahl, W.B.; Patil, K.D.; Kalkwart, K.L. The effect of smoking on the response to periodontal therapy. J. Clin. Periodontol. 1994, 21, 91–97. [Google Scholar] [CrossRef]
- Garcia, R.I. Smokers have less reductions in probing depth than non-smokers following nonsurgical periodontal therapy. Evid.-Based Dent. 2005, 6, 37–38. [Google Scholar] [CrossRef] [Green Version]
- Grossi, S.G.; Zambon, J.; Machtei, E.E.; Schifferle, R.; Andreana, S.; Genco, R.J.; Cummins, D.; Harrap, G. Effects of Smoking and Smoking Cessation on Healing after Mechanical Periodontal Therapy. J. Am. Dent. Assoc. 1997, 128, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kaldahl, W.B.; Johnson, G.K.; Patil, K.D.; Kalkwarf, K.L. Levels of Cigarette Consumption and Response to Periodontal Therapy. J. Periodontol. 1996, 67, 675–681. [Google Scholar] [CrossRef]
- Labriola, A.; Needleman, I.; Moles, D. Systematic review of the effect of smoking on nonsurgical periodontal therapy. Periodontology 2000 2005, 37, 124–137. [Google Scholar] [CrossRef]
- Patel, R.A.; Wilson, R.F.; Palmer, R.M. The Effect of Smoking on Periodontal Bone Regeneration: A Systematic Review and Meta-Analysis. J. Periodontol. 2012, 83, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Paulander, J.; Axelsson, P.; Lindhe, J.; Wennström, J.L. Intra-oral pattern of tooth and periodontal bone loss between the age of 50 and 60 years. A longitudinal prospective study. Acta Odontol. Scand. 2004, 62, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.; Joss, A.; Lang, N.P. Influence of compliance and smoking habits on the outcomes of supportive periodontal therapy (SPT) in a private practice. Oral Health Prev. Dent. 2004, 2, 89–94. [Google Scholar] [PubMed]
- Sculean, A.; Stavropoulos, A.; Berakdar, M.; Windisch, P.; Karring, T.; Brecx, M. Formation of human cementum following different modalities of regenerative therapy. Clin. Oral Investig. 2005, 9, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, A.; Mardas, N.; Herrero, F.; Karring, T. Smoking affects the outcome of guided tissue regeneration with bioresorbable membranes: A retrospective analysis of intrabony defects. J. Clin. Periodontol. 2004, 31, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, M.S. Cigarette Smoking and Periodontal Diseases: Etiology and Management of Disease. Ann. Periodontol. 1998, 3, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.P.; Leung, W.K.; Wong, M.C.M.; Wong, R.M.S.; Wan, P.; Lo, E.C.M.; Corbet, E.F. Effects of smoking on healing response to non-surgical periodontal therapy: A multilevel modelling analysis. J. Clin. Periodontol. 2009, 36, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Bolin, A.; Eklund, G.; Frithiof, L.; Lavstedt, S. The effect of changed smoking habits on marginal alveolar bone loss. A longitudinal study. Swed. Dent. J. 1993, 17, 211–216. [Google Scholar] [PubMed]
- Krall, E.; Dawson-Hughes, B.; Garvey, A.; Garcia, R. Smoking, smoking cessation, and tooth loss. J. Dent. Res. 1997, 76, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergström, J.; Eliasson, S.; Dock, J. A 10-Year Prospective Study of Tobacco Smoking and Periodontal Health. J. Periodontol. 2000, 71, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Rosa, E.F.; Corraini, P.; De Carvalho, V.F.; Inoue, G.; Gomes, E.F.; Lotufo, J.P.B.; De Micheli, G.; Pannuti, C.M. A prospective 12-month study of the effect of smoking cessation on periodontal clinical parameters. J. Clin. Periodontol. 2011, 38, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Duarte, P.M.; Nogueira, C.F.P.; Silva, S.M.; Pannuti, C.M.; Schey, K.C.; Miranda, T.S. Impact of Smoking Cessation on Periodontal Tissues. Int. Dent. J. 2021, 72, 31–36. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Drury, S.; Fu, C.; Schmidt, A.M. Hyperglycemia, glycoxidation and receptor for advanced glycation endproducts: Potential mechanisms underlying diabetic complications, including diabetes-associated periodontitis. Periodontology 2000 2000, 23, 50–62. [Google Scholar] [CrossRef]
- Lalla, E.; Papapanou, P.N. Diabetes mellitus and periodontitis: A tale of two common interrelated diseases. Nat. Rev. Endocrinol. 2011, 7, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Mealey, B.L.; Oates, T.W. American Academy of Periodontology. Diabetes Mellitus and Periodontal Diseases. J. Periodontol. 2006, 77, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Ding, Z.; Yang, Y. The impact of diabetes on periodontal diseases. Periodontology 2000 2020, 82, 214–224. [Google Scholar] [CrossRef]
- Chávarry, N.G.M.; Vettore, M.V.; Sansone, C.; Sheiham, A. The relationship between diabetes mellitus and destructive periodontal disease: A meta-analysis. Oral Health Prev. Dent. 2009, 7, 107–127. [Google Scholar]
- Grossi, S.G.; Genco, R.J. Periodontal Disease and Diabetes Mellitus: A Two-Way Relationship. Ann. Periodontol. 1998, 3, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Lalla, E.; Park, D.B.; Papapanou, P.N.; Lamster, I.B. Oral Disease Burden in Northern Manhattan Patients with Diabetes Mellitus. Am. J. Public Health 2004, 94, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.W.; Burt, B.A.; Becker, M.P.; Genco, R.J.; Shlossman, M. Glycemic Control and Alveolar Bone Loss Progression in Type 2 Diabetes. Ann. Periodontol. 1998, 3, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.W.; Burt, B.A.; Becker, M.P.; Genco, R.J.; Shlossman, M.; Knowler, W.C.; Pettitt, D.J. Severe Periodontitis and Risk for Poor Glycemic Control in Patients with Non-Insulin-Dependent Diabetes Mellitus. J. Periodontol. 1996, 67, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, D.; Marlow, N.M.; Fernandes, J.K.; Leite, R.S. Periodontal disease progression and glycaemic control among Gullah African Americans with type-2 diabetes. J. Clin. Periodontol. 2010, 37, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Guzman, S.; Karima, M.; Wang, H.-Y.; Van Dyke, T.E. Association Between Interleukin-1 Genotype and Periodontal Disease in a Diabetic Population. J. Periodontol. 2003, 74, 1183–1190. [Google Scholar] [CrossRef]
- Seppala, B.; Ainamo, J. A longitudinal study on insulin-dependent diabetes mellitus and periodontal disease. J. Clin. Periodontol. 1993, 20, 161–165. [Google Scholar] [CrossRef]
- Tervonen, T.; Karjalainen, K. Periodontal disease related to diabetic status. A pilot study of the response to periodontal therapy in type 1 diabetes. J. Clin. Periodontol. 1997, 24, 505–510. [Google Scholar] [CrossRef]
- Tervonen, T.; Oliver, R.C. Long-term control of diabetes mellitus and periodontitis. J. Clin. Periodontol. 1993, 20, 431–435. [Google Scholar] [CrossRef]
- Holmlund, A.; Lind, L. Periodontal disease and a poor response to periodontal treatment were associated with an increased risk of incident diabetes: A longitudinal cohort study in Sweden. J. Clin. Periodontol. 2021, 48, 1605–1612. [Google Scholar] [CrossRef]
- Costa, F.O.; Cortelli, J.R.; Cortelli, S.C.; Costa, A.A.; Lima, R.P.E.; Costa, A.M.; Pereira, G.H.M.; Cota, L.O.M. The loss of molars in supportive periodontal care: A 10-year follow-up for tooth- and patient-related factors. J. Clin. Periodontol. 2022, 49, 292–300. [Google Scholar] [CrossRef]
- Westfelt, E.; Rylander, H.; Biohme, G.; Jonasson, P.; Lindhe, J. The effect of periodontal therapy in diabetics. Results after 5 years. J. Clin. Periodontol. 1996, 23, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Christgau, M.; Palitzsch, K.-D.; Schmalz, G.; Kreiner, U.; Frenzel, S. Healing response to non-surgical periodontal therapy in patients with diabetes mellitus: Clinical, microbiological, and immunologic results. J. Clin. Periodontol. 1998, 25, 112–124. [Google Scholar] [CrossRef]
- Faria-Almeida, R.; Navarro, A.; Bascones, A. Clinical and Metabolic Changes After Conventional Treatment of Type 2 Diabetic Patients with Chronic Periodontitis. J. Periodontol. 2006, 77, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Nørskov-Lauritsen, N.; Kilian, M. Reclassification of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, Haemophilus paraphrophilus and Haemophilus segnis as Aggregatibacter actinomycetemcomitans gen. nov., comb. nov., Aggregatibacter aphrophilus comb. nov. and Aggregatibacter segnis comb. nov., and emended description of Aggregatibacter aphrophilus to include V factor-dependent and V factor-independent isolates. Int. J. Syst. Evol. Microbiol. 2006, 56, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, M.; Suzuki, M.; Umeda, M.; Ishikawa, I.; Benno, Y. Reclassification of Bacteroides forsythus (Tanner et al., 1986) as Tannerella forsythensis corrig., gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 841–849. [Google Scholar]
- Maiden, M.F.; Cohee, P.; Tanner, A.C. Proposal to conserve the adjectival form of the specific epithet in the reclassification of Bacteroides forsythus Tanner et al., 1986 to the genus Tannerella Sakamoto et al. 2002 as Tannerella forsythia corrig., gen. nov., comb. nov. Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2003, 53, 2111–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paster, B.J.; Boches, S.K.; Galvin, J.L.; Ericson, R.E.; Lau, C.N.; Levanos, V.A.; Sahasrabudhe, A.; Dewhirst, F.E. Bacterial Diversity in Human Subgingival Plaque. J. Bacteriol. 2001, 183, 3770–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.D.; Koch, G.G.; Rozier, R.G.; Tudor, G.E. Prevalence and Risk Indicators for Periodontal Attachment Loss in a Population of Older Community-Dwelling Blacks and Whites. J. Periodontol. 1990, 61, 521–528. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Socransky, S.S.; Smith, C.; Dibart, S. Relation of baseline microbial parameters to future periodontal attachment loss. J. Clin. Periodontol. 1991, 18, 744–750. [Google Scholar] [CrossRef]
- Grossi, S.G.; Zambon, J.J.; Ho, A.W.; Koch, G.; Dunford, R.G.; Machtei, E.E.; Norderyd, O.M.; Genco, R.J. Assessment of Risk for Periodontal Disease. I. Risk Indicators for Attachment Loss. J. Periodontol. 1994, 65, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Grossi, S.; Genco, R.; Machtet, E.; Ho, A.; Koch, G.; Dunford, R.; Zambon, J.; Hausmann, E. Assessment of Risk for Periodontal Disease. II. Risk Indicators for Alveolar Bone Loss. J. Periodontol. 1995, 66, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Baelum, V.; Luan, W.-M.; Madianos, P.N.; Chen, X.; Fejerskov, O.; Dahlén, G. Subgingival Microbiota in Adult Chinese: Prevalence and Relation to Periodontal Disease Progression. J. Periodontol. 1997, 68, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, M.F.; van der Weijden, G.A.; Abbas, F.; Arief, E.M.; Armand, S.; Winkel, E.G.; van Winkelhoff, A.J.; van der Velden, U. Untreated periodontal disease in Indonesian adolescents. Longitudinal clinical data and prospective clinical and microbiological risk assessment. J. Clin. Periodontol. 2000, 27, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Teanpaisan, R.; Obiechina, N.S.; Pithpornchaiyakul, W.; Pongpaisal, S.; Pisuithanakan, S.; Baelum, V.; Fejerskov, O.; Dahlén, G. Periodontal microbiota and clinical periodontal status in a rural sample in southern Thailand. Eur. J. Oral Sci. 2002, 110, 345–352. [Google Scholar] [CrossRef]
- Fine, D.H.; Markowitz, K.; Furgang, D.; Fairlie, K.; Ferrandiz, J.; Nasri, C.; Mc Kiernan, M.; Gunsolley, J. Aggregatibacter actinomycetemcomitans and Its Relationship to Initiation of Localized Aggressive Periodontitis: Longitudinal Cohort Study of Initially Healthy Adolescents. J. Clin. Microbiol. 2007, 45, 3859–3869. [Google Scholar] [CrossRef] [Green Version]
- Haubek, D.; Ennibi, O.-K.; Poulsen, K.; Væth, M.; Poulsen, S.; Kilian, M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: A prospective longitudinal cohort study. Lancet 2008, 371, 237–242. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Chapple, I.L.C. Biological approaches to the development of novel periodontal therapies—Consensus of the Seventh European Workshop on Periodontology. J. Clin. Periodontol. 2011, 38, 114–118. [Google Scholar] [CrossRef]
- Heitz-Mayfield, L.J.A.; Trombelli, L.; Heitz, F.; Needleman, I.; Moles, D. A systematic review of the effect of surgical debridement vs. non-surgical debridement for the treatment of chronic periodontitis. J. Clin. Periodontol. 2002, 29, 92–102. [Google Scholar] [CrossRef]
- Herrera, D.; Sanz, M.; Jepsen, S.; Needleman, I.; Roldán, S. A systematic review on the effect of systemic antimicrobials as an adjunct to scaling and root planing in periodontitis patients. J. Clin. Periodontol. 2002, 29, 136–159. [Google Scholar] [CrossRef]
- How, K.Y.; Song, K.P.; Chan, K.G. Porphyromonas gingivalis: An Overview of Periodontopathic Pathogen below the Gum Line. Front. Microbiol. 2016, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. Dental biofilms: Difficult therapeutic targets. Periodontology 2000 2002, 28, 12–55. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, L.P. Essential Microbiology for Dentistry, 2nd ed.; Churchil Livingstone Elsevier: London, UK, 2002. [Google Scholar]
- Cutler, C.; Kalmar, J.; Genco, C. Pathogenic strategies of the oral anaerobe, Porphyromonas gingivalis. Trends Microbiol. 1995, 3, 45–51. [Google Scholar] [CrossRef]
- Zeituni, A.E.; Mc Caig, W.; Scisci, E.; Thanassi, D.G.; Cutler, C.W. The Native 67-Kilodalton Minor Fimbria of Porphyromonas gingivalis Is a Novel Glycoprotein with DC-SIGN-Targeting Motifs. J. Bacteriol. 2010, 192, 4103–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzo, P.J.; Cutler, C.W. Microorganisms as risk indicators for periodontal disease. Periodontology 2000 2003, 32, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Zeituni, A.E.; Jotwani, R.; Carrion, J.; Cutler, C.W. Targeting of DC-SIGN on Human Dendritic Cells by Minor Fimbriated Porphyromonas gingivalis Strains Elicits a Distinct Effector T Cell Response. J. Immunol. 2009, 183, 5694–5704. [Google Scholar] [CrossRef] [Green Version]
- Holt, S.C.; Kesavalu, L.; Walker, S.; Genco, C. Virulence factors of Porphyromonas gingivalis. Periodontology 2000 1999, 20, 168–238. [Google Scholar] [CrossRef]
- Hamada, N.; Sojar, H.T.; Cho, M.I.; Genco, R.J. Isolation and characterization of a minor fimbria from Porphyromonas gingivalis. Infect. Immun. 1996, 64, 4788–4794. [Google Scholar] [CrossRef] [Green Version]
- Carrion, J.; Scisci, E.; Miles, B.; Sabino, G.J.; Zeituni, A.E.; Gu, Y.; Bear, A.; Genco, C.A.; Brown, D.L.; Cutler, C.W. Microbial Carriage State of Peripheral Blood Dendritic Cells (DCs) in Chronic Periodontitis Influences DC Differentiation, Atherogenic Potential. J. Immunol. 2012, 189, 3178–3187. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Lamont, R.J. Promoter architecture of the Porphyromonas gingivalis fimbrillin gene. Infect. Immun. 1999, 67, 3227–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Morishima, S.; Takahashi, I.; Hamada, S. Molecular Cloning and Sequencing of the Fimbrilin Gene of Porphyromonas gingivalis Strains and Characterization of Recombinant Proteins. Biochem. Biophys. Res. Commun. 1993, 197, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Kimura, R.K.; Nakamura, T.; Kawabata, S.; Hamada, S. Distribution and molecular characterization of Porphyromonas gingivalis carrying a new type of fimA gene. J. Clin. Microbiol. 2000, 38, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, I.; Amano, A.; Ohara-Nemoto, Y.; Endoh, N.; Morisaki, I.; Kimura, S.; Kawabata, S.; Hamada, S. Identification of a new variant of fimA gene of Porphyromonas gingivalis and its distribution in adults and disabled populations with periodontitis. J. Periodontal Res. 2002, 37, 425–432. [Google Scholar] [CrossRef]
- Genco, C.A.; Van Dyke, T.; Amar, S. Animal models for Porphyromonas gingivalis-mediated periodontal disease. Trends Microbiol. 1998, 6, 444–449. [Google Scholar] [CrossRef]
- Nakano, K.; Kuboniwa, M.; Nakagawa, I.; Yamamura, T.; Nomura, R.; Okahashi, N.; Ooshima, T.; Amano, A. Comparison of inflammatory changes caused by Porphyromonas gingivalis with distinct fimA genotypes in a mouse abscess model. Oral Microbiol. Immunol. 2004, 19, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Neiders, M.E.; Chen, P.B.; Suido, H.; Reynolds, H.S.; Zambon, J.J.; Shlossman, M.; Genco, R.J. Heterogeneity of virulence among strains of Bacteroides gingivalis. J. Periodontal Res. 1989, 24, 192–198. [Google Scholar] [CrossRef]
- Amano, A.; Kuboniwa, A.; Nakagawa, I.; Akiyama, S.; Morisaki, I.; Hamada, S. Prevalence of Specific Genotypes of Porphyromonas gingivalis fimA and Periodontal Health Status. J. Dent. Res. 2000, 79, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Amano, A.; Nakagawa, I.; Kataoka, K.; Morisaki, I.; Hamada, S. Distribution of Porphyromonas gingivalis strains with fimA genotypes in periodontitis patients. J. Clin. Microbiol. 1999, 37, 1426–1430. [Google Scholar] [CrossRef] [Green Version]
- Amano, A.; Nakagawa, I.; Okahashi, N.; Hamada, N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J. Periodontal Res. 2004, 39, 136–142. [Google Scholar] [CrossRef]
- Beikler, T.; Peters, U.; Prajaneh, S.; Prior, K.; Ehmke, B.; Flemmig, T.F. Prevalence of Porphyromonas gingivalis fimA genotypes in Caucasians. Eur. J. Oral Sci. 2003, 111, 390–394. [Google Scholar] [CrossRef]
- Enersen, M.; Olsen, I.; Kvalheim, Ø.; Caugant, D.A. fimA Genotypes and Multilocus Sequence Types of Porphyromonas gingivalis from Patients with Periodontitis. J. Clin. Microbiol. 2008, 46, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missailidis, C.G.; Umeda, J.E.; Ota-Tsuzuki, C.; Anzai, D.; Mayer, M.P.A. Distribution of fimA genotypes of Porphyromonas gingivalis in subjects with various periodontal conditions. Oral Microbiol. Immunol. 2004, 19, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Wang, W.; Zhang, L. The prevalence of fimA genotypes of Porphyromonas gingivalis in patients with chronic periodontitis: A meta-analysis. PLoS ONE 2020, 15, e0240251. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Yoshimura, F.; Duncan, M.J. A regulation cascade controls expression of Porphyromonas gingivalis fimbriae via the FimR response regulator. Mol. Microbiol. 2004, 54, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, X.; Xie, H. Porphyromonas gingivalis short fimbriae are regulated by a FimS/FimR two-component system. FEMS Microbiol. Lett. 2007, 271, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, A.; Sharma, A.; Sojar, H.T.; Kuramitsu, H.K.; Genco, R.J. Effects of temperature stress on expression of fimbriae and superoxide dismutase by Porphyromonas gingivalis. Infect. Immun. 1994, 62, 4682–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Cai, S.; Lamont, R.J. Environmental regulation of fimbrial gene expression in Porphyromonas gingivalis. Infect. Immun. 1997, 65, 2265–2271. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Chung, W.O.; Park, Y.; Lamont, R.J. Regulation of the Porphyromonas gingivalis fimA (Fimbrillin) Gene. Infect. Immun. 2000, 68, 6574–6579. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; James, C.E.; Yoshimura, F.; Lamont, R.J. Expression of the short fimbriae of Porphyromonas gingivalis is regulated in oral bacterial consortia. FEMS Microbiol. Lett. 2006, 262, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.-Y.; Wu, J.; Lamont, R.J.; Lin, X.; Xie, H. Negative Correlation of Distributions of Streptococcus cristatus and Porphyromonas gingivalis in Subgingival Plaque. J. Clin. Microbiol. 2009, 47, 3902–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M. Dendritic cells and the control of immunity: Enhancing the efficiency of antigen presentation. Mt. Sinai J. Med. 2001, 68, 160–166. [Google Scholar]
- Cutler, C.; Jotwani, R. Dendritic Cells at the Oral Mucosal Interface. J. Dent. Res. 2006, 85, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Jotwani, R.; Cutler, C.W. Multiple Dendritic Cell (DC) Subpopulations in Human Gingiva and Association of Mature DCs with CD4+ T-cells in situ. J. Dent. Res. 2003, 82, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Jotwani, R.; Muthukuru, M.; Cutler, C. Increase in HIV Receptors/Co-receptors/α-defensins in Inflamed Human Gingiva. J. Dent. Res. 2004, 83, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Jotwani, R.; Palucka, A.K.; Al-Quotub, M.; Nouri-Shirazi, M.; Kim, J.; Bell, D.; Banchereau, J.; Cutler, C.W. Mature Dendritic Cells Infiltrate the T Cell-Rich Region of Oral Mucosa in Chronic Periodontitis: In Situ, In Vivo, and In Vitro Studies. J. Immunol. 2001, 167, 4693–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Zhang, Y.; Chen, L.; Zhou, T.; Huang, W.; Zhou, X.; Shao, L. The role of Toll-like receptors in periodontitis. Oral Dis. 2016, 23, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Miles, B.; Zakhary, I.; El-Awady, A.; Scisci, E.; Carrion, J.; O’Neill, J.C.; Rawlings, A.; Stern, J.K.; Susin, C.; Cutler, C.W. Secondary Lymphoid Organ Homing Phenotype of Human Myeloid Dendritic Cells Disrupted by an Intracellular Oral Pathogen. Infect. Immun. 2014, 82, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurotaki, D.; Kawase, W.; Sasaki, H.; Nakabayashi, J.; Nishiyama, A.; Morse, H.C.; Ozato, K.; Suzuki, Y.; Tamura, T. Epigenetic control of early dendritic cell lineage specification by the transcription factor IRF8 in mice. Blood 2019, 133, 1803–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, A.; Wu, L. The Early Progenitors of Mouse Dendritic Cells and Plasmacytoid Predendritic Cells Are within the Bone Marrow Hemopoietic Precursors Expressing Flt3. J. Exp. Med. 2003, 198, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstedt, M.; Lundberg, K.; Borrebaeck, C.A.K. Gene Family Clustering Identifies Functionally Associated Subsets of Human In Vivo Blood and Tonsillar Dendritic Cells. J. Immunol. 2005, 175, 4839–4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, K.; Munster, D.J.; Clark, G.J.; Dzionek, A.; Schmitz, J.; Hart, D.N.J. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.R. TGFbeta1 and TNFalpha secreted by mast cells stimulated via the FcepsilonRI activate fibroblasts for high-level production of monocyte chemoattractant protein-1 (MCP-1). Cell Immunol. 2000, 201, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Waskow, C.; Liu, X.; Yao, K.; Hoh, J.; Nussenzweig, M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 2007, 8, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef]
- Meghil, M.; Tawfik, O.K.; Elashiry, M.; Rajendran, M.; Arce, R.M.; Fulton, D.J.; Schoenlein, P.V.; Cutler, C.W. Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front. Immunol. 2019, 10, 2286. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.; Yoshimori, T.; Tooze, S. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- El-Awady, A.R.; Miles, B.; Scisci, E.; Kurago, Z.B.; Palani, C.D.; Arce, R.M.; Waller, J.L.; Genco, C.A.; Slocum, C.; Manning, M.; et al. Porphyromonas gingivalis Evasion of Autophagy and Intracellular Killing by Human Myeloid Dendritic Cells Involves DC-SIGN-TLR2 Crosstalk. PLOS Pathog. 2015, 11, e1004647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, T.L.; Wandel, M.P.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 Targets Damaged Vesicles for Autophagy to Defend Cells against Bacterial Invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Loi, M.; Müller, A.; Steinbach, K.; Niven, J.; da Silva, R.B.; Paul, P.; Ligeon, L.-A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8 + T Cell Responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Quarles, E.; Mekvanich, S.; Kang, A.; Liu, A.; Santos, D.; Miller, R.A.; Rabinovitch, P.S.; Cox, T.C.; Kaeberlein, M. Rapamycin treatment attenuates age-associated periodontitis in mice. GeroScience 2017, 39, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucheryavenko, O.; Nelson, G.; Von Zglinicki, T.; Korolchuk, V.I.; Carroll, B. The mTORC1-autophagy pathway is a target for senescent cell elimination. Biogerontology 2019, 20, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice: I. morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Enk, A.H.; Angeloni, V.L.; Udey, M.C.; Katz, S.I. Inhibition of Langerhans cell antigen-presenting function by IL-10. A role for IL-10 in induction of tolerance. J. Immunol. 1993, 151, 2390–2398. [Google Scholar] [PubMed]
- Simon, J.; Hara, H.; Denfeld, R.; Martin, S. UVB-Irradiated Dendritic Cells Induce Nonproliferating, Regulatory Type T Cells. Ski. Pharmacol. Physiol. 2002, 15, 330–334. [Google Scholar] [CrossRef]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.; Liu, Y.-J.; Mac Pherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Christensen, J.R.; Börnsen, L.; Ratzer, R.; Piehl, F.; Khademi, M.; Olsson, T.; Sørensen, P.S.; Sellebjerg, F. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS ONE 2013, 8, e57820. [Google Scholar] [CrossRef]
- Cutler, C.W.; Jotwani, R.; Palucka, K.A.; Davoust, J.; Bell, D.; Banchereau, J. Evidence and a novel hypothesis for the role of dendritic cells and Porphyromonas gingivalis in adult periodontitis. J. Periodontal Res. 1999, 34, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Cutler, C.W.; Teng, Y.-T.A. Oral mucosal dendritic cells and periodontitis: Many sides of the same coin with new twists. Periodontology 2000 2007, 45, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elashiry, M.; Elashiry, M.M.; Elsayed, R.; Rajendran, M.; Auersvald, C.; Zeitoun, R.; Rashid, M.H.; Ara, R.; Meghil, M.M.; Liu, Y.; et al. Dendritic cell derived exosomes loaded with immunoregulatory cargo reprogram local immune responses and inhibit degenerative bone disease in vivo. J. Extracell. Vesicles 2020, 9, 1795362. [Google Scholar] [CrossRef]
- Steinbrink, K.; Mahnke, K.; Grabbe, S.; Enk, A.H.; Jonuleit, H. Myeloid dendritic cell: From sentinel of immunity to key player of peripheral tolerance? Hum. Immunol. 2009, 70, 289–293. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meghil, M.M.; Ghaly, M.; Cutler, C.W. A Tale of Two Fimbriae: How Invasion of Dendritic Cells by Porphyromonas gingivalis Disrupts DC Maturation and Depolarizes the T-Cell-Mediated Immune Response. Pathogens 2022, 11, 328. https://doi.org/10.3390/pathogens11030328
Meghil MM, Ghaly M, Cutler CW. A Tale of Two Fimbriae: How Invasion of Dendritic Cells by Porphyromonas gingivalis Disrupts DC Maturation and Depolarizes the T-Cell-Mediated Immune Response. Pathogens. 2022; 11(3):328. https://doi.org/10.3390/pathogens11030328
Chicago/Turabian StyleMeghil, Mohamed M., Mira Ghaly, and Christopher W. Cutler. 2022. "A Tale of Two Fimbriae: How Invasion of Dendritic Cells by Porphyromonas gingivalis Disrupts DC Maturation and Depolarizes the T-Cell-Mediated Immune Response" Pathogens 11, no. 3: 328. https://doi.org/10.3390/pathogens11030328
APA StyleMeghil, M. M., Ghaly, M., & Cutler, C. W. (2022). A Tale of Two Fimbriae: How Invasion of Dendritic Cells by Porphyromonas gingivalis Disrupts DC Maturation and Depolarizes the T-Cell-Mediated Immune Response. Pathogens, 11(3), 328. https://doi.org/10.3390/pathogens11030328