
Ecological and Anthropogenic Spatial Gradients Shape Patterns of Dispersal of Foot-and-Mouth Disease Virus in Uganda

 , ,
, ,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Sampling and Sequencing
2.2. Phylogeographic Analysis
2.3. Directional Analyses
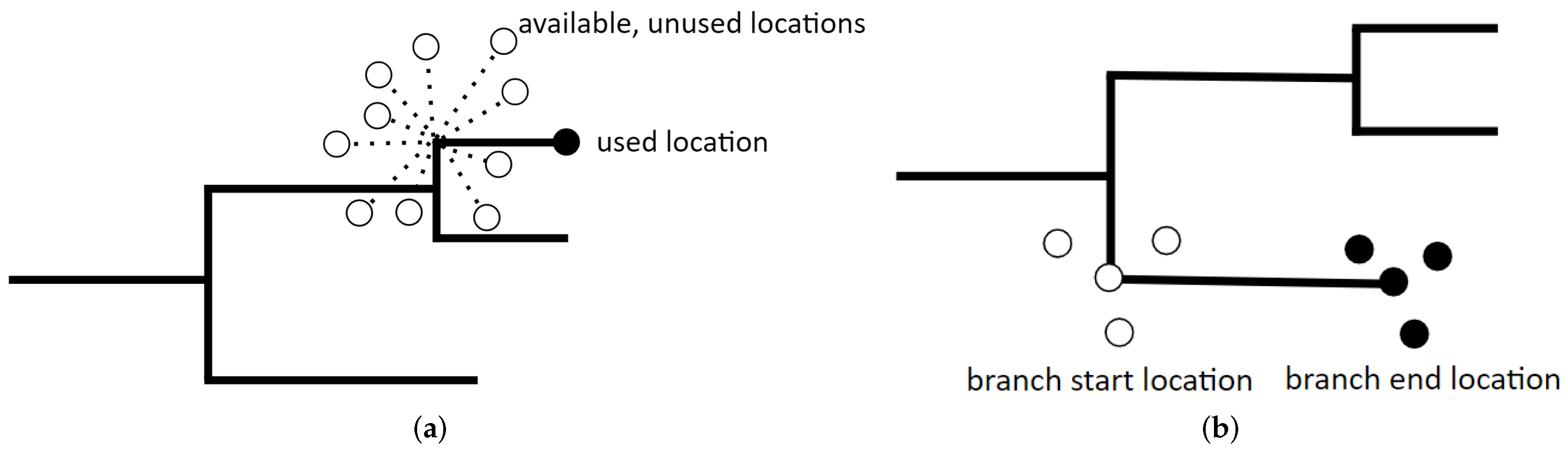
2.4. Step Selection Function Model
2.5. Resource Gradient Function Model
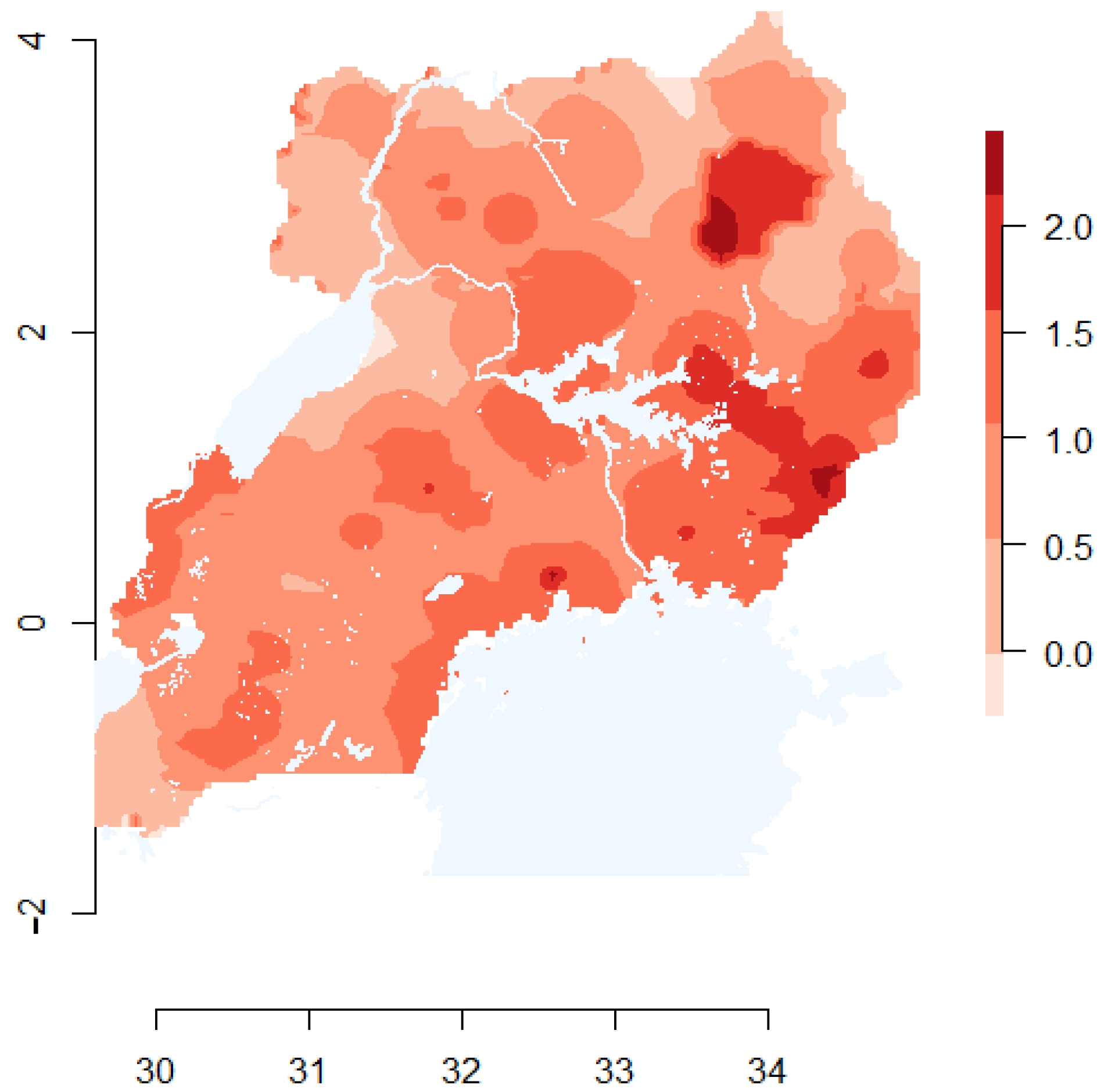
2.6. Viral Source Map
3. Results
3.1. Model Selection and Directional Statistics
3.2. Regression Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus evolution and transmission in an ever more connected world. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellicour, S.; Lequime, S.; Vrancken, B.; Gill, M.S.; Bastide, P.; Gangavarapu, K.; Matteson, N.L.; Tan, Y.; du Plessis, L.; Fisher, A.A.; et al. Epidemiological hypothesis testing using a phylogeographic and phylodynamic framework. Nat. Commun. 2020, 11, 5620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.R.; Shi, W.Q.; Liu, K.; Li, X.L.; Liu, M.J.; Zhang, W.H.; Zhao, G.P.; Chen, J.J.; Zhang, X.A.; Miao, D.; et al. Epidemiology and evolution of Middle East respiratory syndrome coronavirus, 2012–2020. Infect. Dis. Poverty 2021, 10, 66. [Google Scholar] [CrossRef]
- Casey-Bryars, M.; Reeve, R.; Bastola, U.; Knowles, N.J.; Auty, H.; Bachanek-Bankowska, K.; Fowler, V.L.; Fyumagwa, R.; Kazwala, R.; Kibona, T.; et al. Waves of endemic foot-and-mouth disease in eastern Africa suggest feasibility of proactive vaccination approaches. Nat. Ecol. Evol. 2018, 2, 1449–1457. [Google Scholar] [CrossRef]
- Knight-Jones, T.; Rushton, J. The economic impacts of foot and mouth disease—What are they, how big are they and where do they occur? Prev. Vet. Med. 2013, 112, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Perry, B.D.; Rich, K.M. Poverty impacts of foot-and-mouth disease and the poverty reduction implications of its control. Vet. Rec. 2007, 160, 238–241. [Google Scholar] [CrossRef]
- Alexandersen, S.; Mowat, N. Foot-and-mouth disease: Host range and pathogenesis. Foot Mouth Dis. Virus 2005, 288, 9–42. [Google Scholar]
- Munsey, A.; Mwiine, F.N.; Ochwo, S.; Velazquez-Salinas, L.; Ahmed, Z.; Maree, F.; Rodriguez, L.L.; Rieder, E.; Perez, A.; VanderWaal, K. Spatial distribution and risk factors for foot and mouth disease virus in Uganda: Opportunities for strategic surveillance. Prev. Vet. Med. 2019, 171, 104766. [Google Scholar] [CrossRef]
- Munsey, A.; Mwiine, F.N.; Ochwo, S.; Velazquez-Salinas, L.; Ahmed, Z.; Maree, F.; Rodriguez, L.L.; Rieder, E.; Perez, A.; Dellicour, S.; et al. Phylogeographic analysis of foot-and-mouth disease virus serotype O dispersal and associated drivers in East Africa. Mol. Ecol. 2021, 30, 3815–3825. [Google Scholar] [CrossRef]
- Huzurbazar, S. Resource Selection Methods and Applications: Proceedings of the 1st International Conference on Resource Selection, Laramie, Wyoming, 13–15 January 2003; Omnipress: Madison, Wisconsin, 2003. [Google Scholar]
- Manly, B.; McDonald, L.; Thomas, D.; McDonald, T.; Erickson, W. Resource Selection by Animals: Statistical Design and Analysis for Field Studies; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Fortin, D.; Beyer, H.L.; Boyce, M.S.; Smith, D.W.; Duchesne, T.; Mao, J.S. Wolves influence elk movements: Behavior shapes a trophic cascade in Yellowship National Park. Ecology 2005, 86, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Thurfjell, H.; Ciuti, S.; Boyce, M.S. Applications of step-selection functions in ecology and conservation. Mov. Ecol. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avgar, T.; Potts, J.R.; Lewis, M.A.; Boyce, M.S. Integrated step selection analysis: Bridging the gap between resource selection and animal movement. Methods Ecol. Evol. 2016, 7, 619–630. [Google Scholar] [CrossRef]
- Duchesne, T.; Fortin, D.; Rivest, L.P. Equivalence between Step Selection Functions and Biased Correlated Random Walks for Statistical Inference on Animal Movement. PLoS ONE 2015, 10, e0122947. [Google Scholar] [CrossRef]
- Forester, J.D.; Im, H.K.; Rathouz, P.J. Accounting for animal movement in estimation of resource selection functions: Sampling and data analysis. Ecology 2009, 90, 3554–3565. [Google Scholar] [CrossRef]
- Signer, J.; Fieberg, J.; Avgar, T. Estimating utilization distributions from fitted step-selection functions. Ecosphere 2017, 8, e01771. [Google Scholar] [CrossRef]
- Mwiine, F.N.; Velazquez-Salinas, L.; Ahmed, Z.; Ochwo, S.; Munsey, A.; Kenney, M.; Lutwama, J.J.; Maree, F.F.; Lobel, L.; Perez, A.M.; et al. Serological and Phylogenetic Characterization of Foot and Mouth Disease Viruses from Uganda during Cross Sectional Surveillance Study in Cattle between 2014 and 2017. Transbound. Emerg. Dis. 2019, 66, 2011–2024. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Mwiine, F.N.; Ahmed, Z.; Ochwo, S.; Munsey, A.; Lutwama, J.J.; Perez, A.M.; VanderWaal, K.; Rieder, E. Genetic Diversity of Circulating Foot and Mouth Disease Virus in Uganda Cross-Sectional Study During 2014–2017. Front. Vet. Sci. 2020, 7, 162. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Biol. Evol. 2012, 30, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Cambridge, MA, USA, 2015. [Google Scholar] [CrossRef]
- Dellicour, S.; Rose, R.; Faria, N.R.; Lemey, P.; Pybus, O.G. SERAPHIM: Studying environmental rasters and phylogenetically informed movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef]
- Pybus, O.G.; Suchard, M.A.; Lemey, P.; Bernardin, F.J.; Rambaut, A.; Crawford, F.W.; Gray, R.R.; Arinaminpathy, N.; Stramer, S.L.; Busch, M.P.; et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc. Natl. Acad. Sci. USA 2012, 109, 15066–15071. [Google Scholar] [CrossRef] [Green Version]
- Agostinelli, C.; Lund, U. R Package ‘Circular’: Circular Statistics. Available online: https://cran.r-project.org/web/packages/circular/circular.pdf (accessed on 26 April 2022).
- Sanchez-Ramirez, S. rBt: R BEAST Tools (rBt). Available online: https://github.com/santiagosnchez/rBt/blob/master/DESCRIPTION (accessed on 26 April 2022).
- Wang, L.G.; Lam, T.T.Y.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Lam, T.T.Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: A package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Signer, J.; Fieberg, J.; Avgar, T. Animal movement tools (amt): R package for managing tracking data and conducting habitat selection analyses. Ecol. Evol. 2019, 9, 880–890. [Google Scholar] [CrossRef]
- Lele, S.; Keim, J.; Solymos, P. ResourceSelection: Resource Selection (Probability) Functions for Use-Availability Data. Available online: https://cran.r-project.org/web/packages/ResourceSelection/ResourceSelection.pdf (accessed on 26 April 2022).
- Lele, S.R.; Keim, J.L. Weighted distributions and estimation of resource selection probability functions. Ecology 2006, 87, 3021–3028. [Google Scholar] [CrossRef]
- Gail, M.H.; Lubin, J.H.; Rubinstein, L.V. Likelihood calculations for matched case-control studies and survival studies with tied death times. Biometrika 1981, 3, 703–707. [Google Scholar] [CrossRef]
- Logan, J.A. A Multivariate Model for Mobility Tables. Am. J. Sociol. 1983, 2, 324–349. [Google Scholar] [CrossRef]
- Hijmans, R. Raster: Geographic Data Analysis and Modeling. Available online: www.researchgate.net/publication/311921423_Raster_Raster_Geographic_data_analysis_and_modeling (accessed on 26 April 2022).
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Elsevier: Amsterdam, The Netherlands, 1969; pp. 21–132. [Google Scholar] [CrossRef]
- Donaldson, A.I. The Influence of Relative Humidity on the Aerosol Stability of Different Strains of Foot-and-Mouth Disease Virus Suspended in Saliva. J. Gen. Virol. 1972, 15, 25–33. [Google Scholar] [CrossRef]
- Hamoonga, R.; Stevenson, M.; Allepuz, A.; Carpenter, T.; Sinkala, Y. Risk factors for foot-and-mouth disease in Zambia, 1981–2012. Prev. Vet. Med. 2014, 114, 64–71. [Google Scholar] [CrossRef]
- VanderWaal, K.; Gilbertson, M.; Okanga, S.; Allan, B.F.; Craft, M.E. Seasonality and pathogen transmission in pastoral cattle contact networks. R. Soc. Open Sci. 2017, 4, 170808. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, L.M.F.; Santos, L.B.L.; Faria, N.R.; de Castro Silveira, W. Phylogeography of foot-and-mouth disease virus serotype O in Ecuador. Infect. Genet. Evol. 2013, 13, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Bronsvoort, B.M.d.C.; Radford, A.D.; Tanya, V.N.; Nfon, C.; Kitching, R.P.; Morgan, K.L. Molecular Epidemiology of Foot-and-Mouth Disease Viruses in the Adamawa Province of Cameroon. J. Clin. Microbiol. 2004, 42, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motta, P.; Porphyre, T.; Handel, I.G.; Hamman, S.M.; Ngu Ngwa, V.; Tanya, V.N.; Morgan, K.L.; Bronsvoort, B.M.d.C. Characterizing Livestock Markets, Primary Diseases, and Key Management Practices Along the Livestock Supply Chain in Cameroon. Front. Vet. Sci. 2019, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Muleme, M.; Barigye, R.; Khaitsa, M.L.; Berry, E.; Wamono, A.W.; Ayebazibwe, C. Effectiveness of vaccines and vaccination programs for the control of foot-and-mouth disease in Uganda, 2001–2010. Trop. Anim. Health Prod. 2012, 45, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Fournié, G.; Pfeiffer, D.U. Monitoring and controlling disease spread through live animal market networks. Vet. J. 2013, 195, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.J.; Dudík, M.; Elith, J.; Graham, C.H.; Lehmann, A.; Leathwick, J.; Ferrier, S. Sample selection bias and presence-only distribution models: Implications for background and pseudo-absence data. Ecol. Appl. 2009, 19, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Bastos, A.D.S.; Haydon, D.T.; Forsberg, R.; Knowles, N.J.; Anderson, E.C.; Bengis, R.G.; Nel, L.H.; Thomson, G.R. Genetic heterogeneity of SAT-1 type foot-and-mouth disease viruses in southern Africa. Arch. Virol. 2001, 146, 1537–1551. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Anderson, E.C.; Bengis, R.G.; Keet, D.F.; Winterbach, H.K.; Thomson, G.R. Molecular epidemiology of SAT3-type foot-and-mouth disease. Virus Genes 2003, 27, 283–290. [Google Scholar] [CrossRef]
- Samuel, A.R.; Knowles, N.J. Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes). J. Gen. Virol. 2001, 82, 609–621. [Google Scholar] [CrossRef]
- Sobrino, F.; Sáiz, M.; Jiménez-Clavero, M.A.; Núñez, J.I.; Rosas, M.F.; Baranowski, E.; Ley, V. Foot-and-mouth disease virus: A long known virus, but a current threat. Vet. Res. 2001, 32, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Tekleghiorghis, T.; Moormann, R.J.M.; Weerdmeester, K.; Dekker, A. Foot-and-mouth Disease Transmission in Africa: Implications for Control, a Review. Transbound. Emerg. Dis. 2016, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Lasecka-Dykes, L.; Wright, C.F.; Di Nardo, A.; Logan, G.; Mioulet, V.; Jackson, T.; Tuthill, T.J.; Knowles, N.J.; King, D.P. Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa. Viruses 2018, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Variable | OR | 95% CI | p-Value |
|---|---|---|---|
| Low rainfall | 0.42 | 0.22–0.80 | 0.0008 * |
| High rainfall | 0.66 | 0.32–1.34 | 0.253 |
| Low cattle density | 0.22 | 0.06–0.81 | 0.023 * |
| High cattle density | 2.63 | 1.26–5.52 | 0.01 * |
| Near livestock market | 1.88 | 1.05–3.39 | 0.034 * |
| Far from livestock mark | 0.61 | 0.29–1.28 | 0.189 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munsey, A.; Mwiine, F.N.; Ochwo, S.; Velazquez-Salinas, L.; Ahmed, Z.; Rodriguez, L.L.; Rieder, E.; Perez, A.; VanderWaal, K. Ecological and Anthropogenic Spatial Gradients Shape Patterns of Dispersal of Foot-and-Mouth Disease Virus in Uganda. Pathogens 2022, 11, 524. https://doi.org/10.3390/pathogens11050524
Munsey A, Mwiine FN, Ochwo S, Velazquez-Salinas L, Ahmed Z, Rodriguez LL, Rieder E, Perez A, VanderWaal K. Ecological and Anthropogenic Spatial Gradients Shape Patterns of Dispersal of Foot-and-Mouth Disease Virus in Uganda. Pathogens. 2022; 11(5):524. https://doi.org/10.3390/pathogens11050524
Chicago/Turabian StyleMunsey, Anna, Frank Norbert Mwiine, Sylvester Ochwo, Lauro Velazquez-Salinas, Zaheer Ahmed, Luis L. Rodriguez, Elizabeth Rieder, Andres Perez, and Kimberly VanderWaal. 2022. "Ecological and Anthropogenic Spatial Gradients Shape Patterns of Dispersal of Foot-and-Mouth Disease Virus in Uganda" Pathogens 11, no. 5: 524. https://doi.org/10.3390/pathogens11050524
APA StyleMunsey, A., Mwiine, F. N., Ochwo, S., Velazquez-Salinas, L., Ahmed, Z., Rodriguez, L. L., Rieder, E., Perez, A., & VanderWaal, K. (2022). Ecological and Anthropogenic Spatial Gradients Shape Patterns of Dispersal of Foot-and-Mouth Disease Virus in Uganda. Pathogens, 11(5), 524. https://doi.org/10.3390/pathogens11050524