
SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations

 , , ,
, , ,  , , , , ,
, , , , ,  , , ,
, , ,  ,
,  ,
,  , ,
, ,  , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of HCV from Chronically Infected Patients, and of HCV Populations Adapted to Human Hepatoma Cells in Culture
2.2. COVID-19 Patient Cohort, Stratification, and Amplification of SARS-CoV-2 RNA from Diagnostic Samples
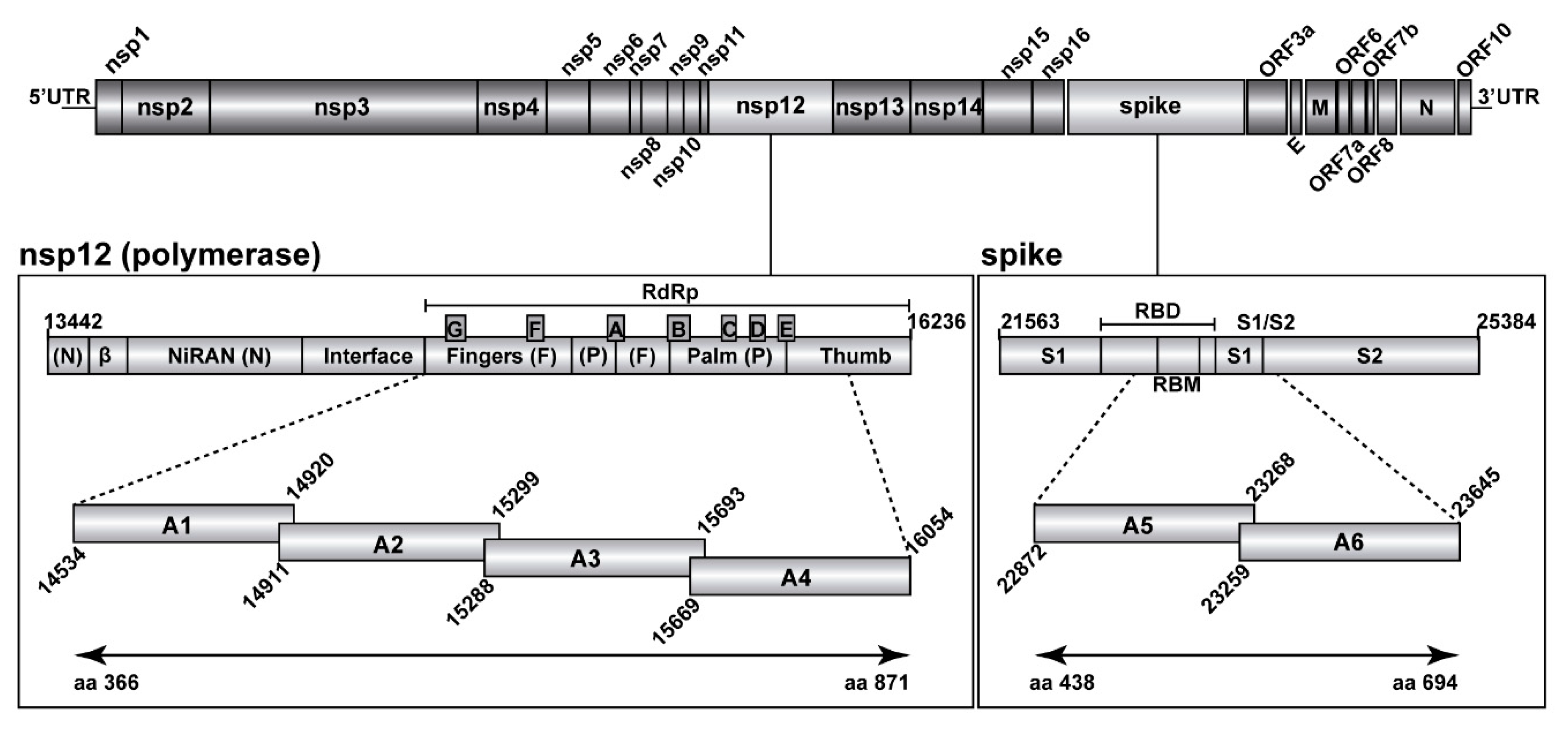
2.3. Ultra-Deep Sequencing of SARS-CoV-2
2.4. Bioinformatics Analyses of SARS-CoV-2 Nucleotide Sequences
2.5. Statistics
3. Results
3.1. A Review of Implications of HCV Population Complexity and Dynamics for Antiviral Resistance and Vaccine Efficacy
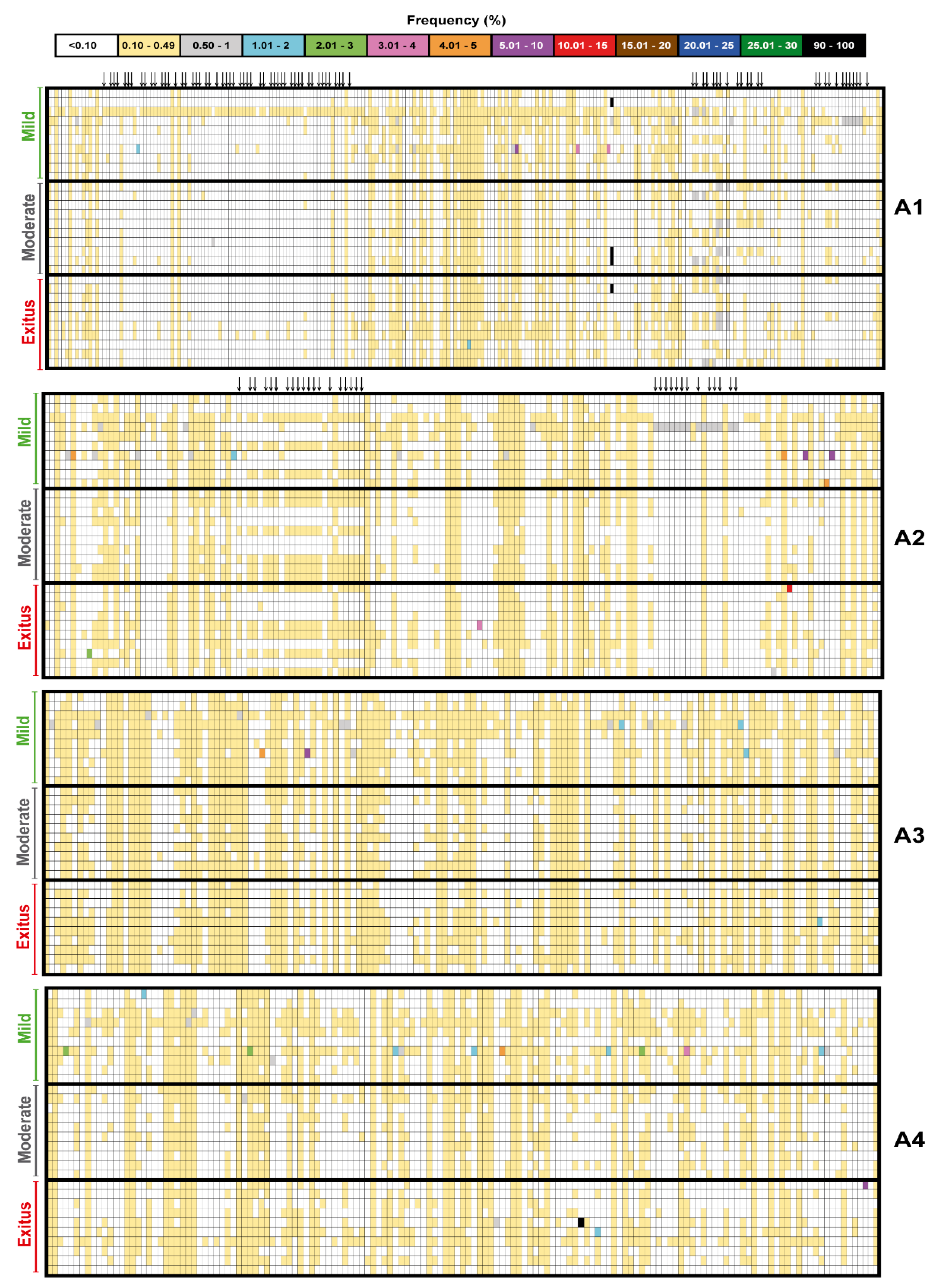
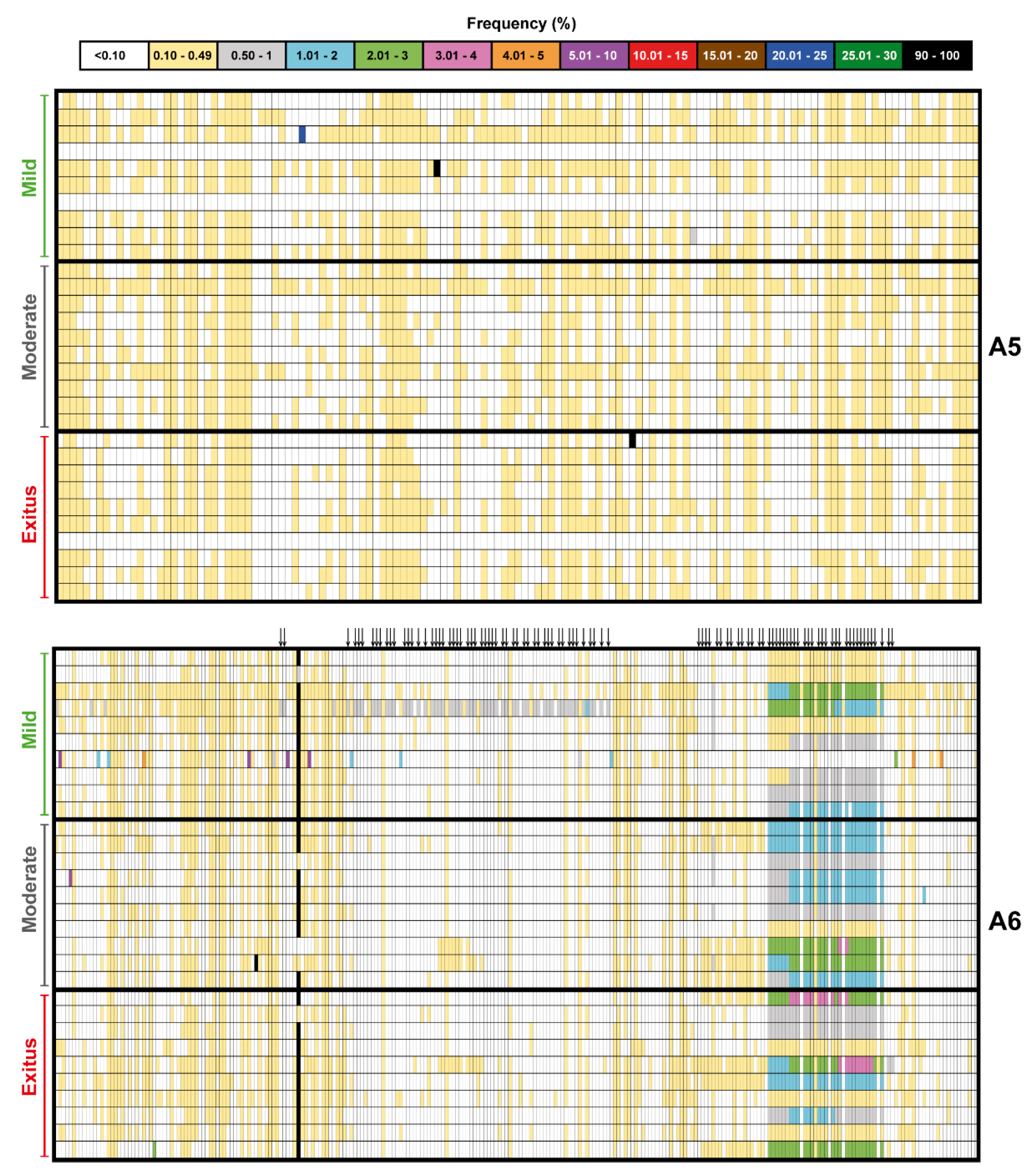
3.2. Ultra-Deep Sequencing Analysis at 0.1% Cut-Off SARS-CoV-2 Mutant Spectra from Patients Progressing towards Different COVID-19 Severity
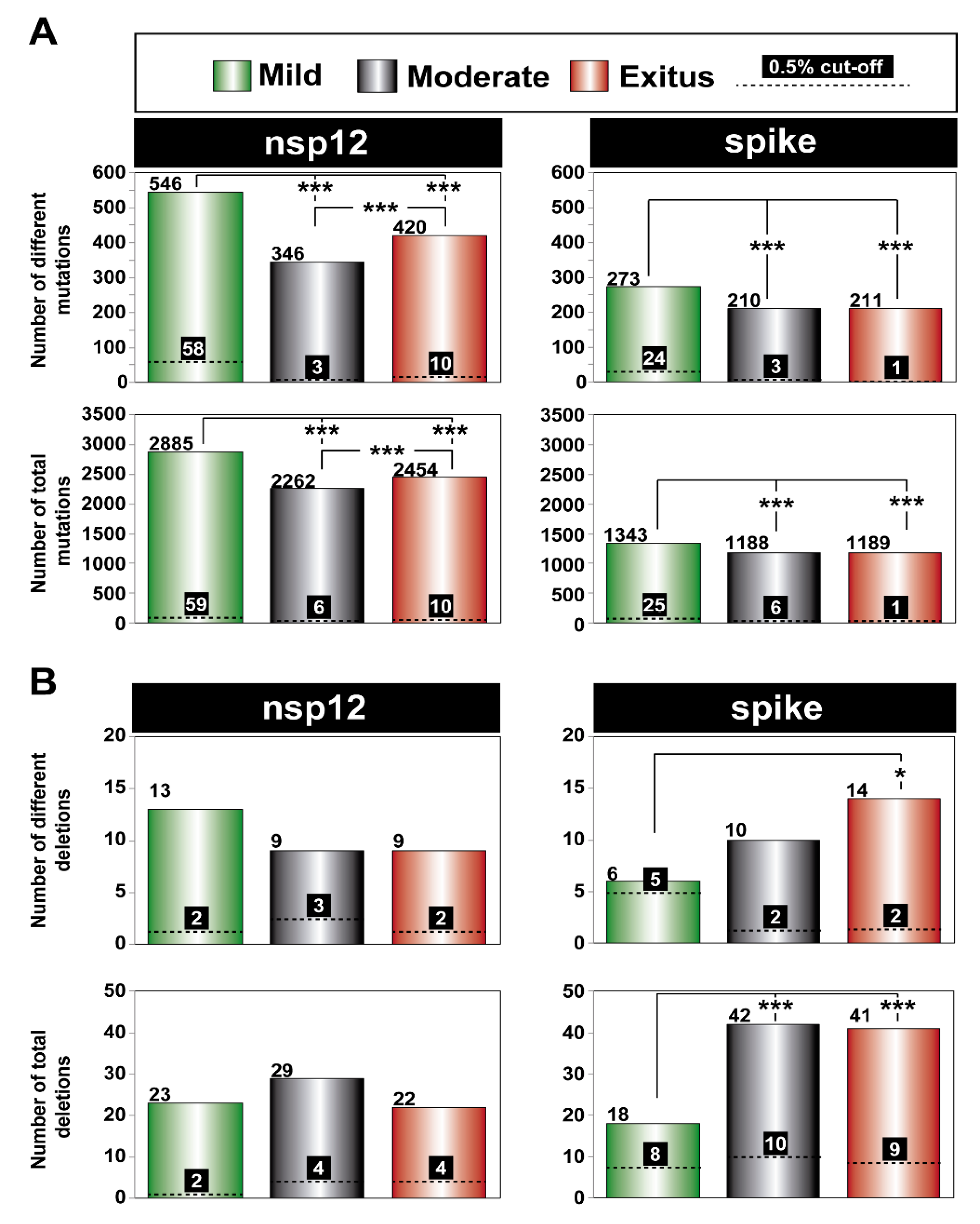
3.3. A Comparison of the SARS-CoV-2 Point Mutation and Deletion Repertoire at 0.5% and 0.1% Frequency Cut-Off
3.4. SARS-CoV-2 Mutation and Deletion Repertoires at Progressively Lower Detection Limit
3.5. Acceptability of the Low Frequency Amino Acid Substitutions
3.6. Comparison of HCV and SARS-CoV-2 Mutant Spectra
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de la Torre, J.C.; Holland, J.J. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J. Virol. 1990, 64, 6278–6281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-López, C.; Arias, A.; Pariente, N.; Gómez-Mariano, G.; Domingo, E. Preextinction viral RNA can interfere with infectivity. J. Virol. 2004, 78, 3319–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirogane, Y.; Watanabe, S.; Yanagi, Y. Cooperation between different RNA virus genomes produces a new phenotype. Nat. Commun. 2012, 3, 1235. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; van Buuren, N.J.; Mateo, R. My Cousin, My Enemy: Quasispecies suppression of drug resistance. Curr. Opin. Virol. 2016, 20, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, K.S.; Hooper, K.A.; Ollodart, A.R.; Dingens, A.S.; Bloom, J.D. Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. eLife 2016, 5, e13974. [Google Scholar] [CrossRef]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [Green Version]
- Grande-Pérez, A.; Lazaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar] [CrossRef] [Green Version]
- Perales, C.; Gallego, I.; de Avila, A.I.; Soria, M.E.; Gregori, J.; Quer, J.; Domingo, E. The increasing impact of lethal mutagenesis of viruses. Future Med. Chem. 2019, 11, 1645–1657. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L. Decoding molnupiravir-induced mutagenesis in SARS-CoV-2. J. Biol. Chem. 2021, 297, 100867. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical perspective on the discovery of the quasispecies concept. Annu. Rev. Virol. 2021, 8, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Buesa-Gomez, J.; Teng, M.N.; Oldstone, C.E.; Oldstone, M.B.; de la Torre, J.C. Variants able to cause growth hormone deficiency syndrome are present within the disease-nil WE strain of lymphocytic choriomeningitis virus. J. Virol. 1996, 70, 8988–8992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, P.I.; Rodriguez, L.L.; Sekellick, M.J. Interferon induction as a quasispecies marker of vesicular stomatitis virus populations. J. Virol. 1998, 72, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; García-Crespo, C.; Soria, M.E.; Perales, C. Viral fitness, population complexity, host interactions, and resistance to antiviral agents. Curr. Top. Microbiol. Immunol. 2022, in press.
- Bull, J.J.; Meyers, L.A.; Lachmann, M. Quasispecies made simple. PLoS Comput. Biol. 2005, 1, e61. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Topfer, A.; Marschall, T.; Bull, R.A.; Luciani, F.; Schonhuth, A.; Beerenwinkel, N. Viral quasispecies assembly via maximal clique enumeration. PLoS Comput. Biol. 2014, 10, e1003515. [Google Scholar] [CrossRef]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Schuster, P. Quasispecies: From Theory to Experimental Systems; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 392. [Google Scholar]
- Ahn, S.; Ke, Z.; Vikalo, H. Viral quasispecies reconstruction via tensor factorization with successive read removal. Bioinformatics 2018, 34, i23–i31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoghegan, J.L.; Holmes, E.C. Evolutionary Virology at 40. Genetics 2018, 210, 1151–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandary, M.B.; Masomian, M.; Poh, C.L. Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. Int. J. Mol. Sci. 2019, 20, 4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henningsson, R.; Moratorio, G.; Borderia, A.V.; Vignuzzi, M.; Fontes, M. DISSEQT-DIStribution-based modeling of SEQuence space Time dynamics. Virus Evol. 2019, 5, vez028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlok, M.; Lang, A.S.; Suttle, C.A. Marine RNA Virus Quasispecies Are Distributed throughout the Oceans. mSphere 2019, 4, e00157-19. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Bessiere, P.; Volmer, R. From one to many: The within-host rise of viral variants. PLoS Pathog. 2021, 17, e1009811. [Google Scholar] [CrossRef]
- Fuhrmann, L.; Jablonski, K.P.; Beerenwinkel, N. Quantitative measures of within-host viral genetic diversity. Curr. Opin. Virol. 2021, 49, 157–163. [Google Scholar] [CrossRef]
- Wu, H.; Liao, B.; Li, X.; Liu, H.; Gong, M.; Shi, H.; Xie, S.; Guo, F.; Chen, K.; Yan, R.; et al. Increased hepatitis B virus quasispecies diversity is correlated with liver fibrosis progression. Infect. Genet. Evol. 2021, 93, 104938. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Z.; Wang, F.S. SARS-associated coronavirus quasispecies in individual patients. N. Engl. J. Med. 2004, 350, 1366–1367. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Huh, H.J.; Kim, Y.J.; Son, D.S.; Jeon, H.J.; Im, E.H.; Kim, J.W.; Lee, N.Y.; Kang, E.S.; Kang, C.I.; et al. Analysis of intrapatient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jary, A.; Leducq, V.; Malet, I.; Marot, S.; Klement-Frutos, E.; Teyssou, E.; Soulie, C.; Abdi, B.; Wirden, M.; Pourcher, V.; et al. Evolution of viral quasispecies during SARS-CoV-2 infection. Clin. Microbiol. Infect. 2020, 26, 1560.e1. [Google Scholar] [CrossRef] [PubMed]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Rueca, M.; Bartolini, B.; Gruber, C.E.M.; Piralla, A.; Baldanti, F.; Giombini, E.; Messina, F.; Marchioni, L.; Ippolito, G.; Di Caro, A.; et al. Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms 2020, 8, 1302. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 exhibits intra-host genomic plasticity and low-frequency polymorphic quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef]
- Khateeb, D.; Gabrieli, T.; Sofer, B.; Hattar, A.; Cordela, S.; Chaouat, A.; Spivak, I.; Lejbkowicz, I.; Almog, R.; Mandelboim, M.; et al. SARS-CoV-2 variants with reduced infectivity and varied sensitivity to the BNT162b2 vaccine are developed during the course of infection. PLoS Pathog. 2022, 18, e1010242. [Google Scholar] [CrossRef]
- Al Khatib, H.A.; Benslimane, F.M.; Elbashir, I.E.; Coyle, P.V.; Al Maslamani, M.A.; Al-Khal, A.; Al Thani, A.A.; Yassine, H.M. Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients with Variable Disease Severities. Front. Cell. Infect. Microbiol. 2020, 10, 575613. [Google Scholar] [CrossRef]
- Nyayanit, D.A.; Yadav, P.D.; Kharde, R.; Shete-Aich, A. Quasispecies analysis of the SARS-CoV-2 from representative clinical samples: A preliminary analysis. Indian J. Med. Res. 2020, 152, 105–107. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef] [PubMed]
- Andres, C.; Garcia-Cehic, D.; Gregori, J.; Pinana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Samarfard, S.; Ramezani, A.; Izadpanah, K.; Afsharifar, A.; Eskandari, M.H.; Karbanowicz, T.P.; Peters, J.R. Quasi-species nature and differential gene expression of severe acute respiratory syndrome coronavirus 2 and phylogenetic analysis of a novel Iranian strain. Infect. Genet. Evol. 2020, 85, 104556. [Google Scholar] [CrossRef]
- Lau, B.T.; Pavlichin, D.; Hooker, A.C.; Almeda, A.; Shin, G.; Chen, J.; Sahoo, M.K.; Huang, C.H.; Pinsky, B.A.; Lee, H.J.; et al. Profiling SARS-CoV-2 mutation fingerprints that range from the viral pangenome to individual infection quasispecies. Genome Med. 2021, 13, 62. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Soria, M.E.; Minguez, P.; Salar-Vidal, L.; Garcia-Crespo, C.; Gallego, I.; Avila, A.; Llorens, C.; Soriano, B.; et al. Vaccine-breakthrough infections with SARS-CoV-2 Alpha mirror mutations in Delta Plus, Iota and Omicron. J. Clin. Investig. 2022, 132, e157700. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, B.; Soria, M.E.; Vazquez-Sirvent, L.; Ferrer-Orta, C.; Lobo-Vega, R.; Minguez, P.; de la Fuente, L.; Llorens, C.; Soriano, B.; Ramos, R.; et al. SARS-CoV-2 Point Mutation and Deletion Spectra and Their Association with Different Disease Outcomes. Microbiol. Spectr. 2022, 10, e0022122. [Google Scholar] [CrossRef]
- Rodriguez-Rivas, J.; Croce, G.; Muscat, M.; Weigt, M. Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes. Proc. Natl. Acad. Sci. USA 2022, 119, e2113118119. [Google Scholar] [CrossRef]
- Garcia-Crespo, C.; Soria, M.E.; Gallego, I.; Avila, A.I.; Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Gomez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. Dissimilar Conservation Pattern in Hepatitis C Virus Mutant Spectra, Consensus Sequences, and Data Banks. J. Clin. Med. 2020, 9, 3450. [Google Scholar] [CrossRef]
- Quer, J.; Rodriguez-Frias, F.; Gregori, J.; Tabernero, D.; Soria, M.E.; Garcia-Cehic, D.; Homs, M.; Bosch, A.; Pinto, R.M.; Esteban, J.I.; et al. Deep sequencing in the management of hepatitis virus infections. Virus Res. 2017, 239, 115–125. [Google Scholar] [CrossRef]
- Perales, C.; Chen, Q.; Soria, M.E.; Gregori, J.; Garcia-Cehic, D.; Nieto-Aponte, L.; Castells, L.; Imaz, A.; Llorens-Revull, M.; Domingo, E.; et al. Baseline hepatitis C virus resistance-associated substitutions present at frequencies lower than 15% may be clinically significant. Infect. Drug Resist. 2018, 11, 2207–2210. [Google Scholar] [CrossRef] [Green Version]
- Soria, M.E.; Gregori, J.; Chen, Q.; Garcia-Cehic, D.; Llorens, M.; de Avila, A.I.; Beach, N.M.; Domingo, E.; Rodriguez-Frias, F.; Buti, M.; et al. Pipeline for specific subtype amplification and drug resistance detection in hepatitis C virus. BMC Infect. Dis. 2018, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Soria, M.E.; Garcia-Crespo, C.; Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Lobo-Vega, R.; de Avila, A.I.; Gallego, I.; Chen, Q.; Garcia-Cehic, D.; Llorens-Revull, M.; et al. Amino Acid Substitutions Associated with Treatment Failure for Hepatitis C Virus Infection. J. Clin. Microbiol. 2020, 58, e01985-20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Perales, C.; Soria, M.E.; Garcia-Cehic, D.; Gregori, J.; Rodriguez-Frias, F.; Buti, M.; Crespo, J.; Calleja, J.L.; Tabernero, D.; et al. Deep-sequencing reveals broad subtype-specific HCV resistance mutations associated with treatment failure. Antiviral. Res. 2020, 174, 104694. [Google Scholar] [CrossRef] [PubMed]
- Quer, J.; Gregori, J.; Rodriguez-Frias, F.; Buti, M.; Madejon, A.; Perez-del-Pulgar, S.; Garcia-Cehic, D.; Casillas, R.; Blasi, M.; Homs, M.; et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J. Clin. Microbiol. 2015, 53, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Perales, C.; Beach, N.M.; Gallego, I.; Soria, M.E.; Quer, J.; Esteban, J.I.; Rice, C.; Domingo, E.; Sheldon, J. Response of hepatitis C virus to long-term passage in the presence of alpha interferon: Multiple mutations and a common phenotype. J. Virol. 2013, 87, 7593–7607. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Gallego, I.; Gregori, J.; Lucia-Sanz, A.; Soria, M.E.; Castro, V.; Beach, N.M.; Manrubia, S.; Quer, J.; Esteban, J.I.; et al. Internal Disequilibria and Phenotypic Diversification during Replication of Hepatitis C Virus in a Noncoevolving Cellular Environment. J. Virol. 2017, 91, e02505-16. [Google Scholar] [CrossRef] [Green Version]
- Gallego, I.; Soria, M.E.; Garcia-Crespo, C.; Chen, Q.; Martinez-Barragan, P.; Khalfaoui, S.; Martinez-Gonzalez, B.; Sanchez-Martin, I.; Palacios-Blanco, I.; de Avila, A.I.; et al. Broad and Dynamic Diversification of Infectious Hepatitis C Virus in a Cell Culture Environment. J. Virol. 2020, 94, e01856-19. [Google Scholar] [CrossRef]
- Domingo, E.; Soria, M.E.; Gallego, I.; de Avila, A.I.; Garcia-Crespo, C.; Martinez-Gonzalez, B.; Gomez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. A new implication of quasispecies dynamics: Broad virus diversification in absence of external perturbations. Infect. Genet. Evol. 2020, 82, 104278. [Google Scholar] [CrossRef]
- Delgado, S.; Perales, C.; Garcia-Crespo, C.; Soria, M.E.; Gallego, I.; de Avila, A.I.; Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Lopez-Galindez, C.; Moran, F.; et al. A Two-Level, Intramutant Spectrum Haplotype Profile of Hepatitis C Virus Revealed by Self-Organized Maps. Microbiol. Spectr. 2021, 9, e0145921. [Google Scholar] [CrossRef]
- Soria, M.E.; Corton, M.; Martinez-Gonzalez, B.; Lobo-Vega, R.; Vazquez-Sirvent, L.; Lopez-Rodriguez, R.; Almoguera, B.; Mahillo, I.; Minguez, P.; Herrero, A.; et al. High SARS-CoV-2 viral load is associated with a worse clinical outcome of COVID-19 disease. Access Microbiol. 2021, 3, 000259. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, N.J.; Parobek, C.M.; Juliano, J.J.; Bailey, J.A. SeekDeep: Single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res. 2018, 46, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.F.; Doolittle, R.F. Progressive alignment of amino acid sequences and construction of phylogenetic trees from them. Methods Enzymol. 1996, 266, 368–382. [Google Scholar]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. S8), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, V.C.; Cento, V.; Lenci, I.; Aragri, M.; Rossi, P.; Barbaliscia, S.; Melis, M.; Verucchi, G.; Magni, C.F.; Teti, E.; et al. Multiclass HCV resistance to direct-acting antiviral failure in real-life patients advocates for tailored second-line therapies. Liver Int. 2017, 37, 514–528. [Google Scholar] [CrossRef]
- Dietz, J.; Susser, S.; Vermehren, J.; Peiffer, K.H.; Grammatikos, G.; Berger, A.; Ferenci, P.; Buti, M.; Mullhaupt, B.; Hunyady, B.; et al. Patterns of Resistance-Associated Substitutions in Patients with Chronic HCV Infection Following Treatment with Direct-Acting Antivirals. Gastroenterology 2018, 154, 976–988.e974. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R.; Pianko, S.; Brown, A.; Forton, D.; Nahass, R.G.; George, J.; Barnes, E.; Brainard, D.M.; Massetto, B.; Lin, M.; et al. Efficacy of sofosbuvir plus ribavirin with or without peginterferon-alfa in patients with hepatitis C virus genotype 3 infection and treatment-experienced patients with cirrhosis and hepatitis C virus genotype 2 infection. Gastroenterology 2015, 149, 1462–1470. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M.; et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [Green Version]
- Lawitz, E.; Lalezari, J.P.; Hassanein, T.; Kowdley, K.V.; Poordad, F.F.; Sheikh, A.M.; Afdhal, N.H.; Bernstein, D.E.; Dejesus, E.; Freilich, B.; et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: A randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 2013, 13, 401–408. [Google Scholar] [CrossRef]
- Lawitz, E.; Gane, E.J. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013, 369, 678–679. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Maekawa, S.; Komatsu, N.; Tatsumi, A.; Miura, M.; Muraoka, M.; Suzuki, Y.; Amemiya, F.; Takano, S.; Fukasawa, M.; et al. Deep sequencing and phylogenetic analysis of variants resistant to interferon-based protease inhibitor therapy in chronic hepatitis induced by genotype 1b hepatitis C virus. J. Virol. 2015, 89, 6105–6116. [Google Scholar] [CrossRef] [Green Version]
- Stross, C.; Shimakami, T.; Haselow, K.; Ahmad, M.Q.; Zeuzem, S.; Lange, C.M.; Welsch, C. Natural HCV variants with increased replicative fitness due to NS3 helicase mutations in the C-terminal helix alpha18. Sci. Rep. 2016, 6, 19526. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.C.; De Meyer, S.; Bartels, D.J.; Dierynck, I.; Zhang, E.Z.; Spanks, J.; Tigges, A.M.; Ghys, A.; Dorrian, J.; Adda, N.; et al. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin. Infect. Dis. 2013, 57, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Svarovskaia, E.S.; Dvory-Sobol, H.; Parkin, N.; Hebner, C.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Xu, S.; Ku, K.; et al. Infrequent development of resistance in genotype 1-6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin. Infect. Dis. 2014, 59, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, J.; Beach, N.M.; Moreno, E.; Gallego, I.; Pineiro, D.; Martinez-Salas, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; et al. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J. Virol. 2014, 88, 12098–12111. [Google Scholar] [CrossRef] [Green Version]
- Gallego, I.; Gregori, J.; Soria, M.E.; Garcia-Crespo, C.; Garcia-Alvarez, M.; Gomez-Gonzalez, A.; Valiergue, R.; Gomez, J.; Esteban, J.I.; Quer, J.; et al. Resistance of high fitness hepatitis C virus to lethal mutagenesis. Virology 2018, 523, 100–109. [Google Scholar] [CrossRef]
- Gallego, I.; Sheldon, J.; Moreno, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Barrier-Independent, Fitness-Associated Differences in Sofosbuvir Efficacy against Hepatitis C Virus. Antimicrob. Agents Chemother. 2016, 60, 3786–3793. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; de Avila, A.I.; Gallego, I.; Sheldon, J.; Perales, C. Viral fitness: History and relevance for viral pathogenesis and antiviral interventions. Pathog. Dis. 2019, 77, ftz021. [Google Scholar] [CrossRef]
- Domingo, E.; Elena, S.F.; Perales, C.; Schuster, P. Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Delgado, S.; Moran, F.; Mora, A.; Merelo, J.J.; Briones, C. A novel representation of genomic sequences for taxonomic clustering and visualization by means of self-organizing maps. Bioinformatics 2015, 31, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Kohonen, T. Self-Organizing Maps; Springer: Berlin/Heidelberg, Germany, 2001; Volume 501. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Perales, C. Quasispecies dynamics and clinical significance of hepatitis C virus (HCV) antiviral resistance. Int. J. Antimicrob. Agents 2018, 56, 105562. [Google Scholar] [CrossRef]
- Wong, Y.C.; Lau, S.Y.; Wang To, K.K.; Mok, B.W.Y.; Li, X.; Wang, P.; Deng, S.; Woo, K.F.; Du, Z.; Li, C.; et al. Natural Transmission of Bat-like Severe Acute Respiratory Syndrome Coronavirus 2 Without Proline-Arginine-Arginine-Alanine Variants in Coronavirus Disease 2019 Patients. Clin. Infect. Dis. 2021, 73, e437–e444. [Google Scholar] [CrossRef]
- Tang, J.W.; Cheung, J.L.; Chu, I.M.; Sung, J.J.; Peiris, M.; Chan, P.K. The large 386-nt deletion in SARS-associated coronavirus: Evidence for quasispecies? J. Infect. Dis. 2006, 194, 808–813. [Google Scholar] [CrossRef]
- Liu, J.; Lim, S.L.; Ruan, Y.; Ling, A.E.; Ng, L.F.; Drosten, C.; Liu, E.T.; Stanton, L.W.; Hibberd, M.L. SARS transmission pattern in Singapore reassessed by viral sequence variation analysis. PLoS Med. 2005, 2, e43. [Google Scholar] [CrossRef] [Green Version]
- Borucki, M.K.; Lao, V.; Hwang, M.; Gardner, S.; Adney, D.; Munster, V.; Bowen, R.; Allen, J.E. Middle East Respiratory Syndrome Coronavirus Intra-Host Populations Are Characterized by Numerous High Frequency Variants. PLoS ONE 2016, 11, e0146251. [Google Scholar] [CrossRef]
- Posthuma, C.C.; Te Velthuis, A.J.W.; Snijder, E.J. Nidovirus RNA polymerases: Complex enzymes handling exceptional RNA genomes. Virus Res. 2017, 234, 58–73. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Duerr, R.; Dimartino, D.; Marier, C.; Zappile, P.; Wang, G.; Plitnick, J.; Griesemer, S.B.; Lasek-Nesselquist, E.; Dittmann, M.; Ortigoza, M.B.; et al. Delta-Omicron recombinant SARS-CoV-2 in a transplant patient treated with Sotrovimab. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gu, H.; Ng, D.Y.M.; Liu, G.Y.Z.; Cheng, S.S.M.; Krishnan, P.; Chang, L.D.J.; Cheuk, S.S.Y.; Hui, M.M.Y.; Lam, T.T.Y.; Peiris, M.; et al. Recombinant BA.1/BA.2 SARS-CoV-2 Virus in Arriving Travelers, Hong Kong, February 2022. Emerg. Infect. Dis. 2022, 28, 1276–1278. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, e00408–e00420. [Google Scholar] [CrossRef]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, D.; Lon, J.R.; Chen, X.; Hu, M.; Lin, S.; Chen, Z.; Wang, X.; Meng, Y.; Du, H. Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 100, 164–173. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Acciarri, C.; Galli, M.; Zehender, G. Early phylogenetic estimate of the effective reproduction number of SARS-CoV-2. J. Med. Virol. 2020, 92, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Nabil, B.; Sabrina, B.; Abdelhakim, B. Transmission route and introduction of pandemic SARS-CoV-2 between China, Italy, and Spain. J. Med. Virol. 2021, 93, 564–568. [Google Scholar] [CrossRef]
- Pereson, M.J.; Mojsiejczuk, L.; Martinez, A.P.; Flichman, D.M.; Garcia, G.H.; Di Lello, F.A. Phylogenetic analysis of SARS-CoV-2 in the first few months since its emergence. J. Med. Virol. 2021, 93, 1722–1731. [Google Scholar] [CrossRef]
- Castells, M.; Lopez-Tort, F.; Colina, R.; Cristina, J. Evidence of increasing diversification of emerging Severe Acute Respiratory Syndrome Coronavirus 2 strains. J. Med. Virol. 2020, 92, 2165–2172. [Google Scholar] [CrossRef]
- Diez-Fuertes, F.; Iglesias-Caballero, M.; Garcia-Perez, J.; Monzon, S.; Jimenez, P.; Varona, S.; Cuesta, I.; Zaballos, A.; Jimenez, M.; Checa, L.; et al. A Founder Effect Led Early SARS-CoV-2 Transmission in Spain. J. Virol. 2021, 95, e01583-20. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, S.; Shi, C.M.; Song, S.; Zhu, S.; Su, Y.; Zhao, W.; Li, M.; Bao, Y.; Xue, Y.; et al. Population Genetics of SARS-CoV-2: Disentangling Effects of Sampling Bias and Infection Clusters. Genom. Proteom. Bioinform. 2020, 18, 640–647. [Google Scholar] [CrossRef]
- Domingo, E. Virus as Populations, 2nd ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Smith, E.C.; Denison, M.R. Coronaviruses as DNA wannabes: A new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013, 9, e1003760. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Garcia-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogando, N.S.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Bredenbeek, P.J.; Posthuma, C.C.; Snijder, E.J. The Enzymatic Activity of the nsp14 Exoribonuclease Is Critical for Replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020, 94, e01246-20. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell. Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef]
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuan, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef] [Green Version]
- Aaskov, J.; Buzacott, K.; Thu, H.M.; Lowry, K.; Holmes, E.C. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 2006, 311, 236–238. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Category | ||||||
|---|---|---|---|---|---|---|
| Total | Mild | Moderate | Exitus | |||
| nsp12 (polymerase) | Number of different mutations | Transitions (Ts) (%) | 578 (98.97%) | 544 (99.63%) | 344 (99.42%) | 416 (99.05%) |
| Transversions (Tv) (%) | 6 (1.03%) | 2 (0.37%) | 2 (0.58%) | 4 (0.24%) | ||
| Ratio (Ts/Tv) | 96.33 | 272 | 172 | 104 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| Number of total mutations | Transitions (Ts) (%) | 7587 (99.82%) | 2883 (99.93%) | 2254 (99.65%) | 2451 (99.84%) | |
| Transversions (Tv) (%) | 14 (0.18%) | 2 (0.07%) | 8 (0.35%) | 4 (0.16%) | ||
| Ratio (Ts/Tv) | 541.93 | 1441.50 | 281.75 | 612.75 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| spike | Number of different mutations | Transitions (Ts) (%) | 297 (99.33%) | 273 (100%) | 209 (99.52%) | 210 (99.53%) |
| Transversions (Tv) (%) | 2 (0.67%) | 0 (0%) | 1 (0.48%) | 1 (0.47%) | ||
| Ratio (Ts/Tv) | 148.50 | - | 209 | 210 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| Number of total mutations | Transitions (%) | 3718 (99.95%) | 1343 (100%) | 1187 (99.92%) | 1188 (99.92%) | |
| Transversions (%) | 2 (0.05%) | 0 (0%) | 1 (0.08%) | 1 (0.08%) | ||
| Ratio (Ts/Tv) | 1859 | - | 1187 | 1188 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| Patient Category | ||||||
|---|---|---|---|---|---|---|
| Total | Mild | Moderate | Exitus | |||
| nsp12 (polymerase) | Number of different mutations | Synonymous (Syn) (%) | 238 (40.75%) | 218 (39.93%) | 146 (42.20%) | 175 (41.67%) |
| Non-synonymous (Non-syn) (%) | 346 (59.25%) | 328 (60.07%) | 200 (57.80%) | 245 (58.33%) | ||
| Ratio (Syn/Non-syn) | 0.69 | 0.66 | 0.73 | 0.71 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| Number of total mutations | Synonymous (Syn) (%) | 2971 (39.08%) | 1130 (39.17%) | 877 (38.78%) | 964 (39.27%) | |
| Non-synonymous (Non-syn) (%) | 4631 (60.92%) | 1755 (60.83%) | 1385 (61.23%) | 1491 (60.73%) | ||
| Ratio (Syn/Non-syn) | 0.64 | 0.64 | 0.63 | 0.65 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
| spike | Number of different mutations | Synonymous (Syn) (%) | 125 (41.95%) | 115 (42.28%) | 90 (43.06%) | 89 (42.38%) |
| Non-synonymous (Non-syn) (%) | 173 (58.05%) | 157 (57.72%) | 119 (56.94%) | 121 (57.62%) | ||
| Ratio (Syn/Non-syn) | 0.72 | 0.73 | 0.76 | 0.74 | ||
| p-value | <0.001 | <0.001 | 0.006 | 0.002 | ||
| Significance a | *** | *** | *** | *** | ||
| Number of total mutations | Synonymous (Syn) (%) | 1659 (44.60%) | 606 (45.12%) | 525 (44.19%) | 528 (44.41%) | |
| Non-synonymous (Non-syn) (%) | 2061 (55.40%) | 737 (54.88%) | 663 (55.81%) | 661 (55.59%) | ||
| Ratio (Syn/Non-syn) | 0.80 | 0.82 | 0.79 | 0.80 | ||
| p-value | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Significance a | *** | *** | *** | *** | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-González, B.; Soria, M.E.; Vázquez-Sirvent, L.; Ferrer-Orta, C.; Lobo-Vega, R.; Mínguez, P.; de la Fuente, L.; Llorens, C.; Soriano, B.; Ramos-Ruíz, R.; et al. SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations. Pathogens 2022, 11, 662. https://doi.org/10.3390/pathogens11060662
Martínez-González B, Soria ME, Vázquez-Sirvent L, Ferrer-Orta C, Lobo-Vega R, Mínguez P, de la Fuente L, Llorens C, Soriano B, Ramos-Ruíz R, et al. SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations. Pathogens. 2022; 11(6):662. https://doi.org/10.3390/pathogens11060662
Chicago/Turabian StyleMartínez-González, Brenda, María Eugenia Soria, Lucía Vázquez-Sirvent, Cristina Ferrer-Orta, Rebeca Lobo-Vega, Pablo Mínguez, Lorena de la Fuente, Carlos Llorens, Beatriz Soriano, Ricardo Ramos-Ruíz, and et al. 2022. "SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations" Pathogens 11, no. 6: 662. https://doi.org/10.3390/pathogens11060662
APA StyleMartínez-González, B., Soria, M. E., Vázquez-Sirvent, L., Ferrer-Orta, C., Lobo-Vega, R., Mínguez, P., de la Fuente, L., Llorens, C., Soriano, B., Ramos-Ruíz, R., Cortón, M., López-Rodríguez, R., García-Crespo, C., Somovilla, P., Durán-Pastor, A., Gallego, I., de Ávila, A. I., Delgado, S., Morán, F., ... Perales, C. (2022). SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations. Pathogens, 11(6), 662. https://doi.org/10.3390/pathogens11060662