
Genome Analysis of Klebsiella pneumoniae Reveals International High-Risk Pandemic MDR Clones Emerging in Tertiary Healthcare Settings in Uganda

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites
2.2. Bacteria Isolation
2.3. Antimicrobial Susceptibility Test
2.4. Selection Criteria
2.5. Library Preparation
2.6. Sequence Quality Check, Assembly, and Taxonomic Assignment
2.7. Genome Sequence Analysis
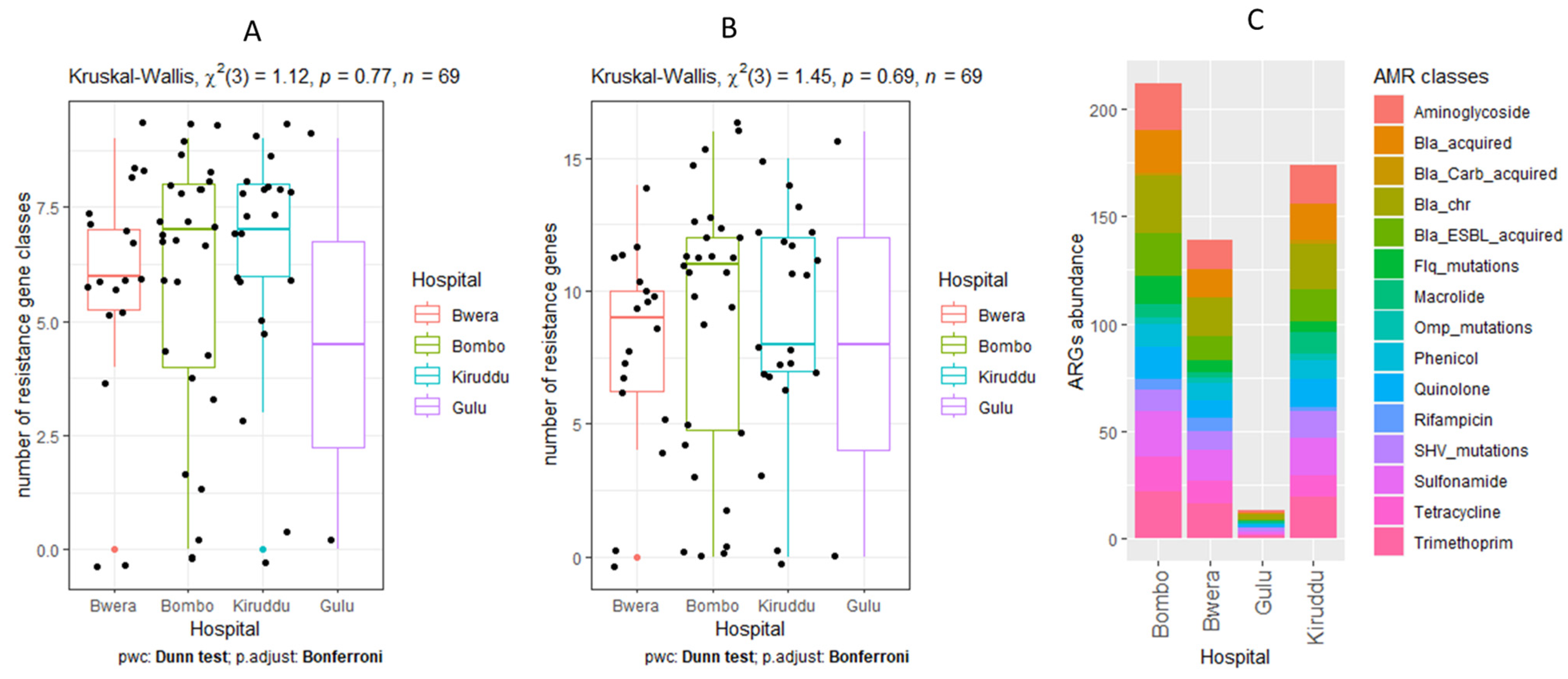
2.8. Statistical Analysis
3. Results
3.1. Characteristics of the MDR Isolates Used in the Study
3.2. Antibiotic Susceptibility of the Isolates
3.3. Klebsiella Sequence Types (STs), K-Serotype, and O-Serotype
3.4. Clonal Groups
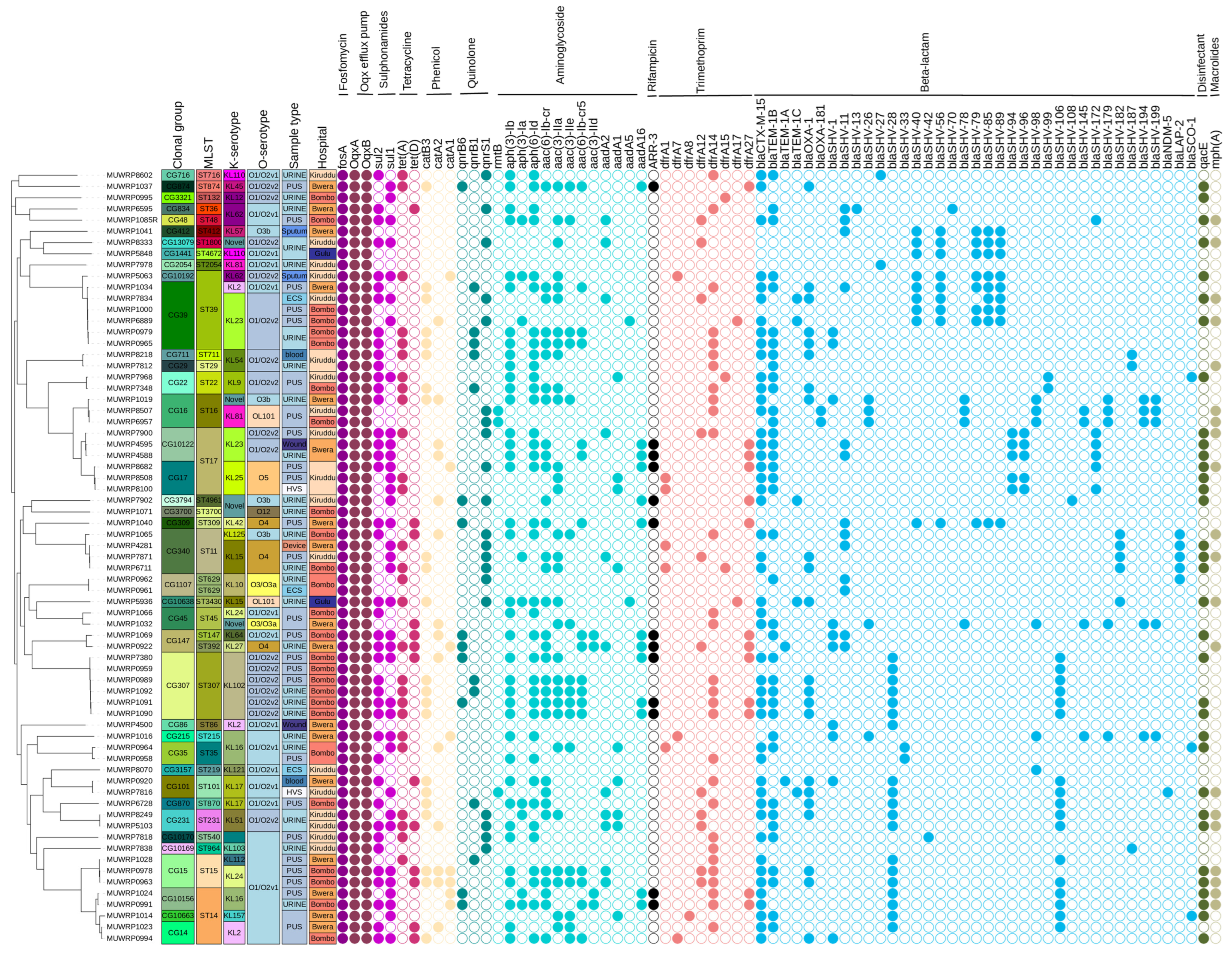
3.5. Detection of AMR Genes
3.6. Mutations Associated with Antibiotic Resistance
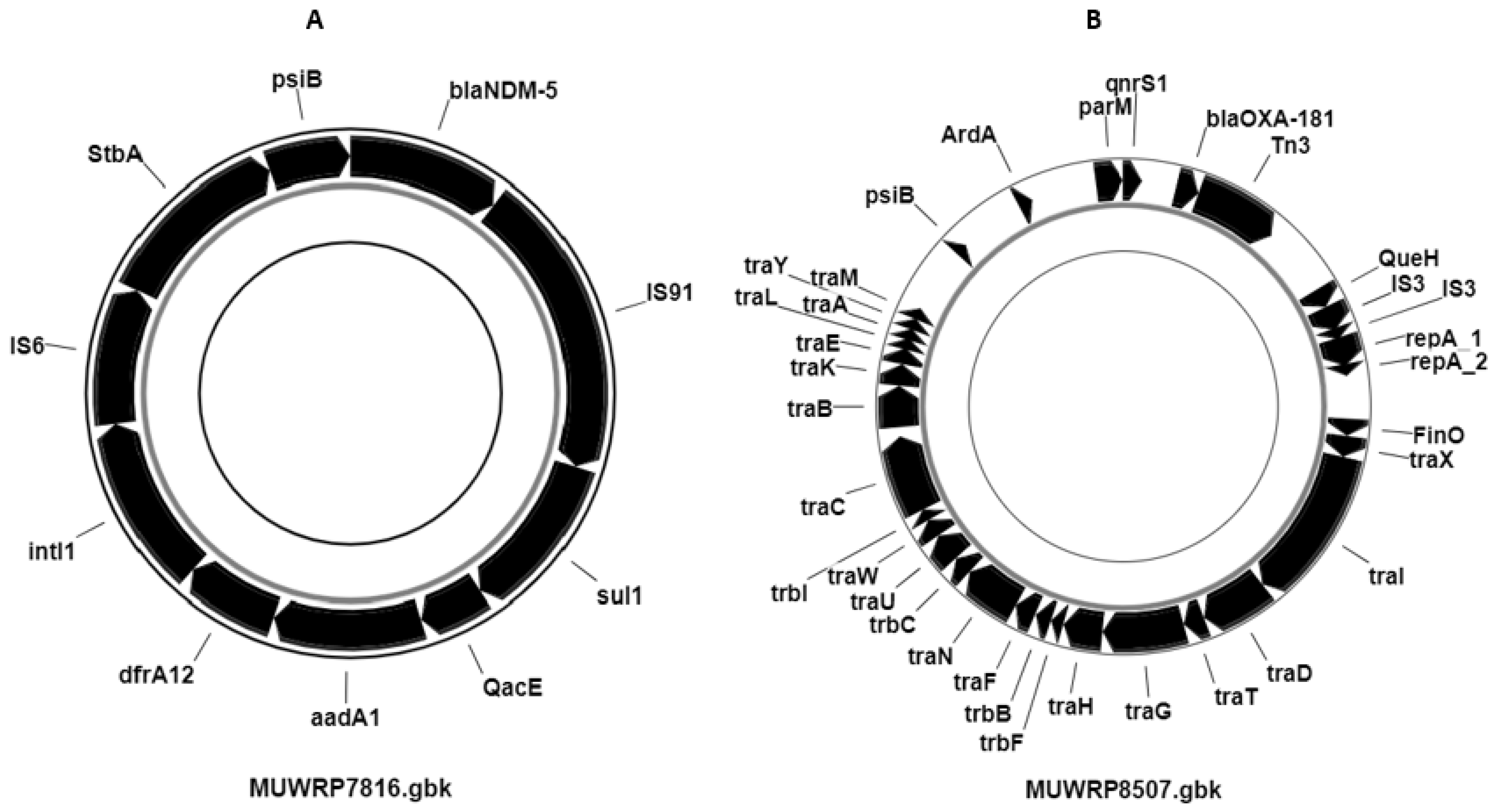
3.7. Distribution of Plasmids and Other Mobile Genetic Elements
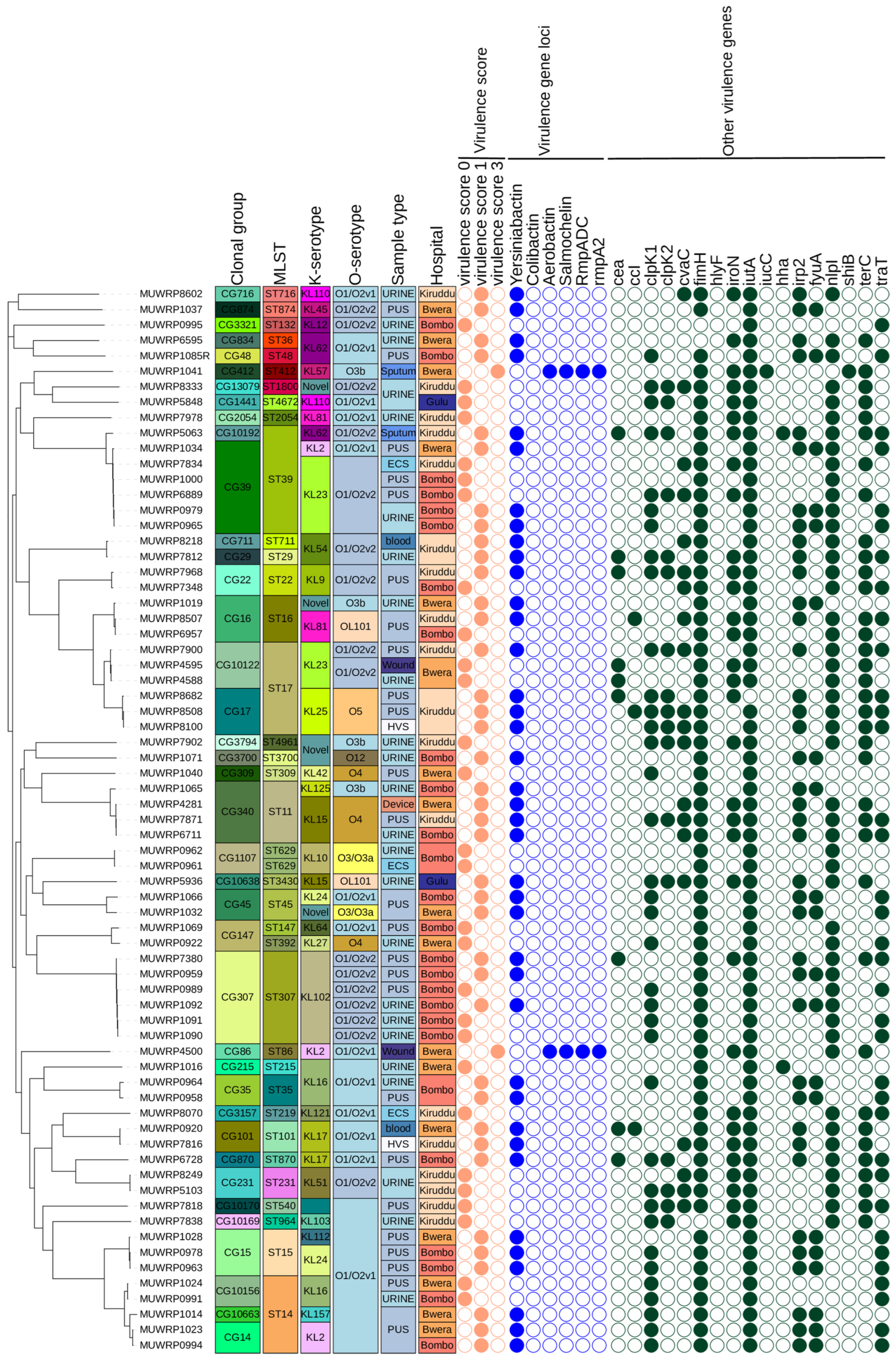
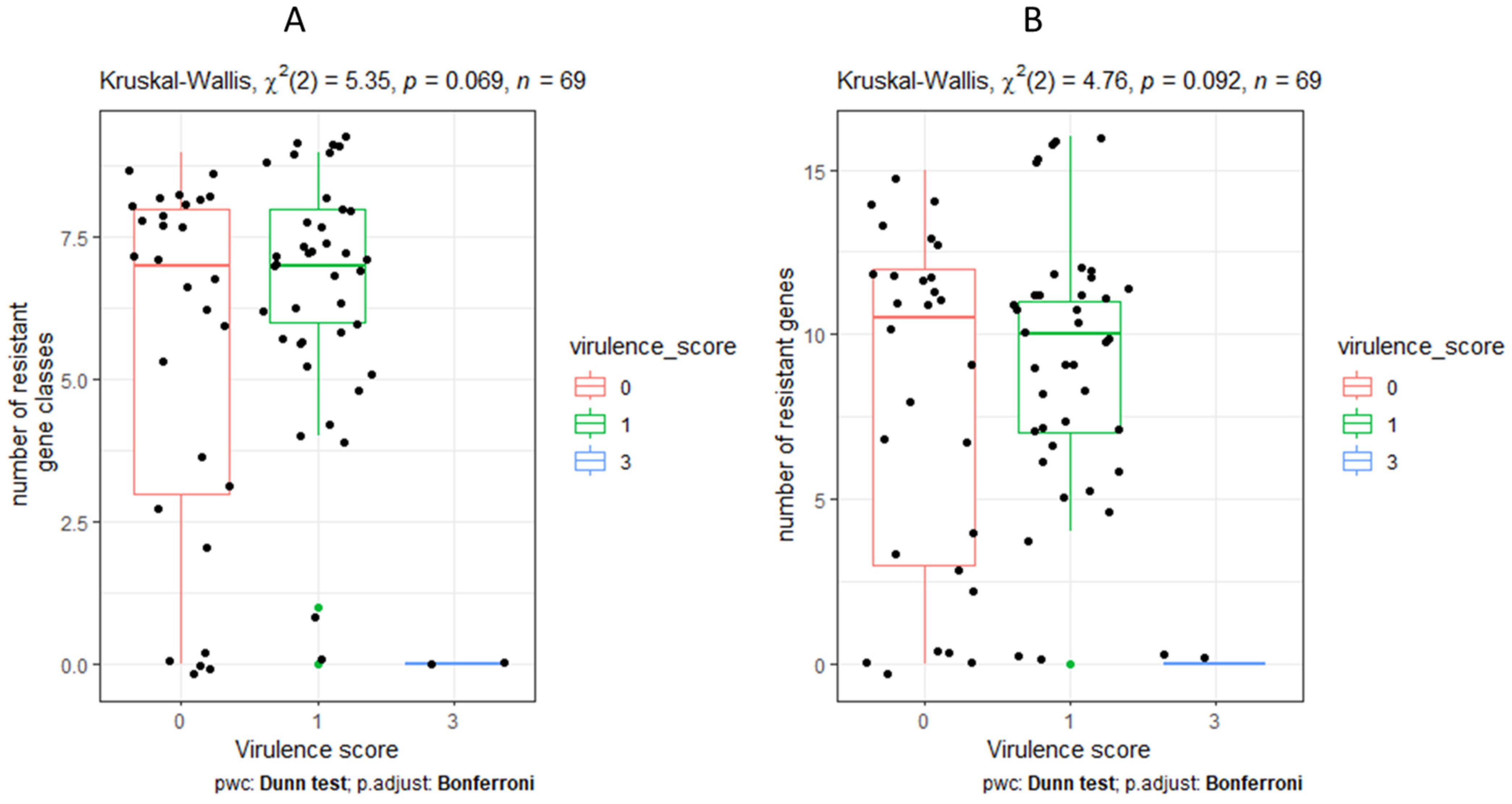
3.8. Characteristics and Distribution of Virulent Genes
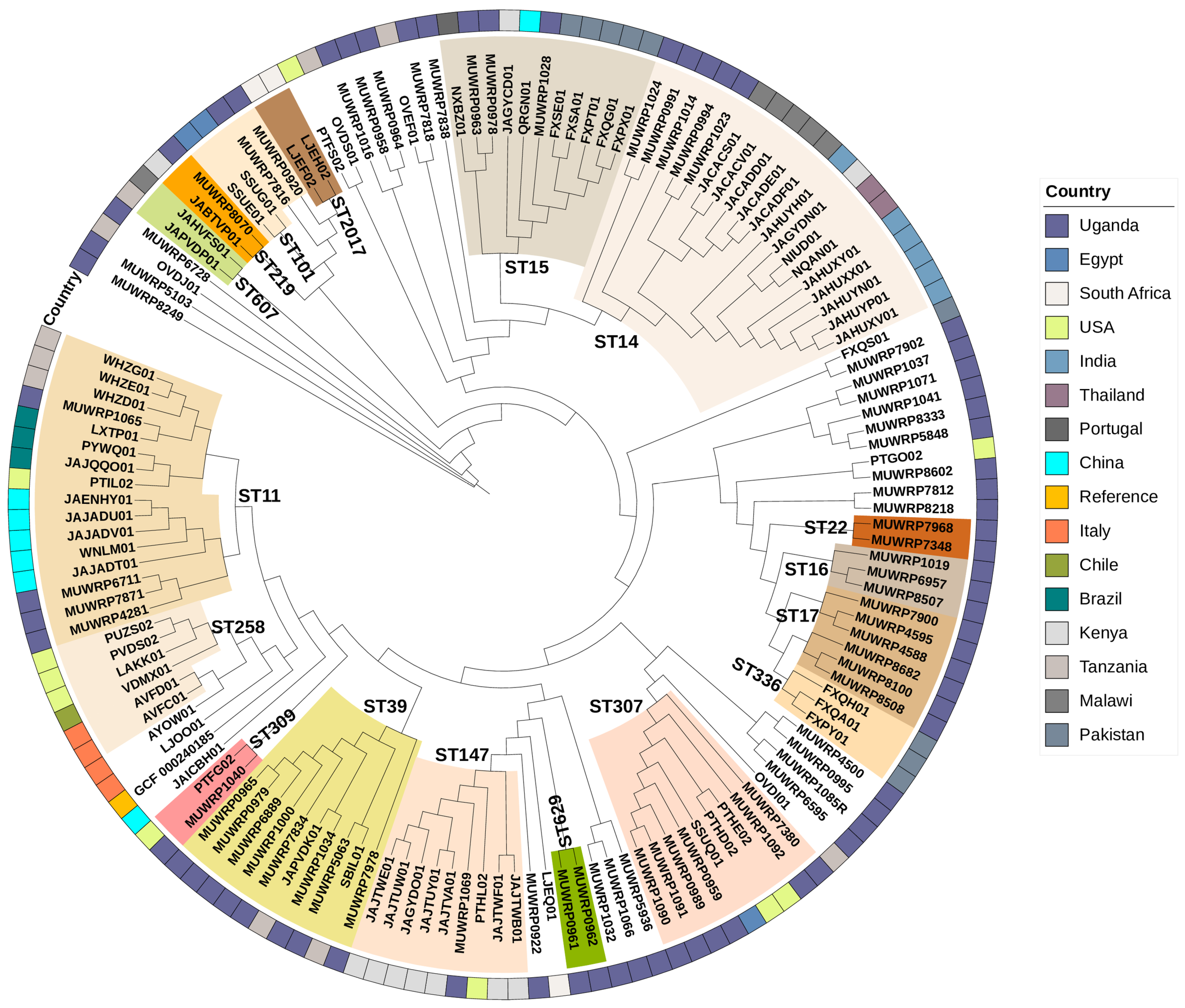
3.9. Phylogenetic Relationships
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef] [PubMed]
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO global priority pathogens list: A bibliometric analysis of medline-pubmed for knowledge mobilization to infection prevention and control practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Bialek-Davenet, S.; Criscuolo, A.; Ailloud, F.; Passet, V.; Jones, L.; Delannoy-Vieillard, A.S.; Garin, B.; Le Hello, S.; Arlet, G.; Nicolas-Chanoine, M.H.; et al. Genomic definition of hypervirulent and multidrug-resistant klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 2014, 20, 1812–1820. [Google Scholar] [CrossRef]
- Shon, A.S.; Bajwa, R.P.S.; Russo, T.A. Hypervirulent (hypermucoviscous) Klebsiella Pneumoniae: A new and dangerous breed. Virulence 2013, 4, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Du, P.; Xiao, N.; Ji, F.; Russo, T.A.; Guo, J. Hypervirulent Klebsiella pneumoniae is emerging as an increasingly prevalent K. pneumoniae pathotype responsible for nosocomial and healthcare-associated infections in Beijing, China. Virulence 2020, 11, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Chang, H.Y.; Lai, Y.C.; Pan, C.C.; Tsai, S.F.; Peng, H.L. Sequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43. Gene 2004, 337, 189–198. [Google Scholar] [CrossRef]
- Liu, Y.C.; Cheng, D.L.; Lin, C.L. Klebsiella Pneumoniae Liver Abscess Associated with Septic Endophthalmitis [Internet]. Available online: http://archinte.jamanetwork.com/ (accessed on 8 August 2023).
- Chen, L.; Kreiswirth, B.N. Convergence of carbapenem-resistance and hypervirulence in Klebsiella pneumoniae. In The Lancet Infectious Diseases; Lancet Publishing Group: New York, NY, USA, 2018; Volume 18, pp. 2–3. [Google Scholar]
- Arcari, G.; Carattoli, A. Global spread and evolutionary convergence of multidrug-resistant and hypervirulent Klebsiella pneumoniae high-risk clones. In Pathogens and Global Health; Taylor and Francis Ltd.: Boca Raton, FL, USA, 2023; Volume 117, pp. 328–341. [Google Scholar]
- Russo, T.A.; Marr, C.M. Hypervirulent Klebsiella pneumoniae. Clin. Microbiol. Rev. 2019, 32, 1110–1128. [Google Scholar] [CrossRef]
- Schaufler, K.; Nowak, K.; Düx, A.; Semmler, T.; Villa, L.; Kourouma, L.; Bangoura, K.; Wieler, L.H.; Leendertz, F.H.; Guenther, S. Clinically relevant ESBL-producing K. pneumoniae ST307 and E. coli ST38 in an urban West African rat population. Front. Microbiol. 2018, 9, 150. [Google Scholar] [CrossRef]
- Muraya, A.; Kyany’a, C.; Kiyaga, S.; Smith, H.J.; Kibet, C.; Martin, M.J.; Kimani, J.; Musila, L. Antimicrobial Resistance and Virulence Characteristics of Klebsiella pneumoniae Isolates in Kenya by Whole-Genome Sequencing. Pathogens 2022, 11, 545. [Google Scholar] [CrossRef]
- Ssekatawa, K.; Byarugaba, D.K.; Nakavuma, J.L.; Kato, C.D.; Ejobi, F.; Tweyongyere, R.; Eddie, W.M. Prevalence of pathogenic Klebsiella pneumoniae based on PCR capsular typing harbouring carbapenemases encoding genes in Uganda tertiary hospitals. Antimicrob. Resist. Infect. Control 2021, 10, 57. [Google Scholar] [CrossRef]
- Struve, C.; Roe, C.C.; Stegger, M.; Stahlhut, S.G.; Hansen, D.S.; Engelthaler, D.M.; Andersen, P.S.; Driebe, E.M.; Keim, P.; Krogfelt, K.A. Mapping the evolution of hypervirulent Klebsiella pneumoniae. mBio 2015, 6, e00630-15. [Google Scholar] [CrossRef] [PubMed]
- Wyres, K.L.; Nguyen, T.N.T.; Lam, M.M.C.; Judd, L.M.; Van Vinh Chau, N.; Dance, D.A.B.; Ip, M.; Karkey, A.; Ling, C.L.; Miliya, T.; et al. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med. 2020, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Hounmanou, Y.M.G.; Wanyana, A.; Alafi, S.; Wabwire-Mangen, F.; Christensen, H.; Olsen, J.E.; Byarugaba, D.K. Whole strains vs MGEs in short and longterm transmission of ESBL genes between healthcare and community settings in Uganda. Sci. Rep. 2023, 13, 10229. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, CLSI Supplement M100, 33rd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2023. [Google Scholar]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Kong, Y. Btrim: A fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 2011, 98, 152–153. [Google Scholar] [CrossRef]
- Nederbragt, A.J. On the middle ground between open source and commercial software-the case of the Newbler program. In Genome Biology; BioMed Central Ltd.: London, UK, 2014; Volume 15. [Google Scholar]
- Wyres, K.L.; Wick, R.R.; Gorrie, C.; Jenney, A.; Follador, R.; Thomson, N.R.; Holt, K.E. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. 2016, 2, e000102. [Google Scholar] [CrossRef]
- Lam, M.M.C.; Wick, R.R.; Watts, S.C.; Cerdeira, L.T.; Wyres, K.L.; Holt, K.E. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 2021, 12, 4188. [Google Scholar] [CrossRef]
- Hennart, M.; Guglielmini, J.; Bridel, S.; Maiden, M.C.J.; Jolley, K.A.; Criscuolo, A.; Brisse, S. A Dual Barcoding Approach to Bacterial Strain Nomenclature: Genomic Taxonomy of Klebsiella pneumoniae Strains. Mol. Biol. Evol. 2022, 39, msac135. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Rogers, M.R.C.; Braat, J.C.; Verschuuren, T.D.; Top, J.; Corander, J.; Willems, R.J.; Schürch, A.C. mlplasmids: A user-friendly tool to predict plasmid-and chromosome-derived sequences for single species. Microb. Genom. 2018, 4, e000224. [Google Scholar] [CrossRef]
- Tanizawa, Y.; Fujisawa, T.; Kaminuma, E.; Nakamura, Y.; Arita, M. DFAST and DAGA: Web-based integrated genome annotation tools and resources. Biosci. Microbiota Food Health 2016, 35, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2018, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Bertels, F.; Silander, O.K.; Pachkov, M.; Rainey, P.B.; Van Nimwegen, E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.r-project.org/ (accessed on 14 August 2021).
- Wyres, K.L.; Wick, R.R.; Judd, L.M.; Froumine, R.; Tokolyi, A.; Gorrie, C.L.; Lam, M.M.C.; Duchêne, S.; Jenney, A.; Holt, K.E. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019, 15, e1008114. [Google Scholar] [CrossRef]
- Hussain, A.; Mazumder, R.; Ahmed, A.; Saima, U.; Phelan, J.E.; Campino, S.; Ahmed, D.; Asadulghani; Clark, T.G.; Mondal, D. Genome dynamics of high-risk resistant and hypervirulent Klebsiella pneumoniae clones in Dhaka, Bangladesh. Front. Microbiol. 2023, 14, 1184196. [Google Scholar] [CrossRef]
- Lowe, M.; Kock, M.M.; Coetzee, J.; Hoosien, E.; Peirano, G.; Strydom, K.A.; Ehlers, M.M.; Mbelle, N.M.; Shashkina, E.; Haslam, D.B.; et al. Klebsiella pneumoniae ST307 with blaoxa-181, South Africa, 2014–2016. Emerg. Infect. Dis. 2019, 25, 739–747. [Google Scholar] [CrossRef]
- Loconsole, D.; Accogli, M.; De Robertis, A.L.; Capozzi, L.; Bianco, A.; Morea, A.; Mallamaci, R.; Quarto, M.; Parisi, A.; Chironna, M. Emerging high-risk ST101 and ST307 carbapenem-resistant Klebsiella pneumoniae clones from bloodstream infections in Southern Italy. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 24. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Rudin, S.D.; Marshall, S.H.; Coakley, P.; Chen, L.; Kreiswirth, B.N.; Rather, P.N.; Hujer, A.M.; Toltzis, P.; van Duin, D.; et al. OqxAB, a quinolone and olaquindox efflux pump, is widely distributed among multidrug-resistant Klebsiella pneumoniae isolates of human origin. Antimicrob. Agents Chemother. 2013, 57, 4602–4603. [Google Scholar] [CrossRef] [PubMed]
- Magobo, R.E.; Ismail, H.; Lowe, M.; Strasheim, W.; Mogokotleng, R.; Perovic, O.; Kwenda, S.; Ismail, A.; Makua, M.; Bore, A.; et al. Outbreak of NDM-1- and OXA-181-Producing Klebsiella pneumoniae Bloodstream Infections in a Neonatal Unit, South Africa. Emerg. Infect. Dis. 2023, 29, 1531–1539. Available online: http://www.ncbi.nlm.nih.gov/pubmed/37486166 (accessed on 13 September 2023). [CrossRef] [PubMed]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef]
- Ampaire, L.; Katawera, V.; Nyehangane, D.; Boum, Y.; Bazira, J. Epidemiology of Carbapenem Resistance among Multi-drug Resistant Enterobacteriaceae in Uganda. Br. Microbiol. Res. J. 2015, 8, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Usman Qamar, M.; SLopes, B.; Hassan, B.; Khurshid, M.; Shafique, M.; Atif Nisar, M.; Mohsin, M.; Nawaz, Z.; Muzammil, S.; Aslam, B.; et al. The present danger of New Delhi metallo-β-lactamase: A threat to public health. Future Microbiol. 2020, 15, 1759–1778. [Google Scholar] [CrossRef]
- Wu, K.M.; Li, N.H.; Yan, J.J.; Tsao, N.; Liao, T.L.; Tsai, H.C.; Fung, C.P.; Chen, H.J.; Liu, Y.M.; Wang, J.T.; et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J. Bacteriol. 2009, 191, 4492–4501. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byarugaba, D.K.; Erima, B.; Wokorach, G.; Alafi, S.; Kibuuka, H.; Mworozi, E.; Najjuka, F.; Kiyengo, J.; Musinguzi, A.K.; Wabwire-Mangen, F. Genome Analysis of Klebsiella pneumoniae Reveals International High-Risk Pandemic MDR Clones Emerging in Tertiary Healthcare Settings in Uganda. Pathogens 2023, 12, 1334. https://doi.org/10.3390/pathogens12111334
Byarugaba DK, Erima B, Wokorach G, Alafi S, Kibuuka H, Mworozi E, Najjuka F, Kiyengo J, Musinguzi AK, Wabwire-Mangen F. Genome Analysis of Klebsiella pneumoniae Reveals International High-Risk Pandemic MDR Clones Emerging in Tertiary Healthcare Settings in Uganda. Pathogens. 2023; 12(11):1334. https://doi.org/10.3390/pathogens12111334
Chicago/Turabian StyleByarugaba, Denis K., Bernard Erima, Godfrey Wokorach, Stephen Alafi, Hannah Kibuuka, Edison Mworozi, Florence Najjuka, James Kiyengo, Ambrose K. Musinguzi, and Fred Wabwire-Mangen. 2023. "Genome Analysis of Klebsiella pneumoniae Reveals International High-Risk Pandemic MDR Clones Emerging in Tertiary Healthcare Settings in Uganda" Pathogens 12, no. 11: 1334. https://doi.org/10.3390/pathogens12111334
APA StyleByarugaba, D. K., Erima, B., Wokorach, G., Alafi, S., Kibuuka, H., Mworozi, E., Najjuka, F., Kiyengo, J., Musinguzi, A. K., & Wabwire-Mangen, F. (2023). Genome Analysis of Klebsiella pneumoniae Reveals International High-Risk Pandemic MDR Clones Emerging in Tertiary Healthcare Settings in Uganda. Pathogens, 12(11), 1334. https://doi.org/10.3390/pathogens12111334