
Upregulation of Neuropilin-1 Inhibits HTLV-1 Infection
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
2.4. Western Blot Analysis
2.5. Flow Cytometry
2.6. Chromatin Immunoprecipitation (ChIP) Assays
2.7. Small RNA Interference
2.8. Transfection and Single-Cycle, Replication-Dependent Infection Assays
2.9. Lentiviral Transduction
2.10. Isolation of Virions and Detection of Gag p19 in Culture Media
2.11. In Silico Analysis and Statistical Analysis
3. Results
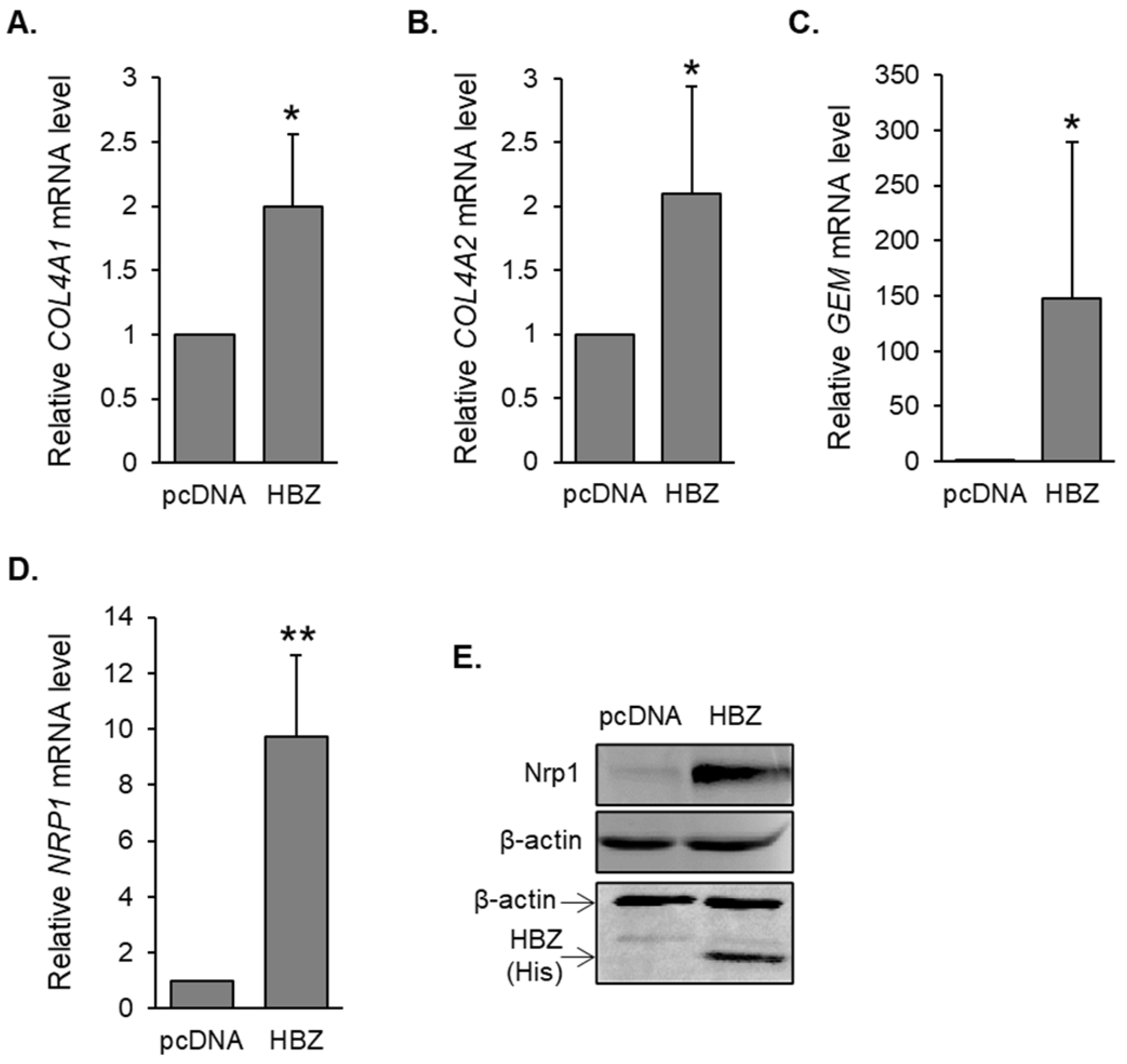
3.1. Genes Involved in HTLV-1 Infection Are Upregulated in HBZ-Expressing Cells
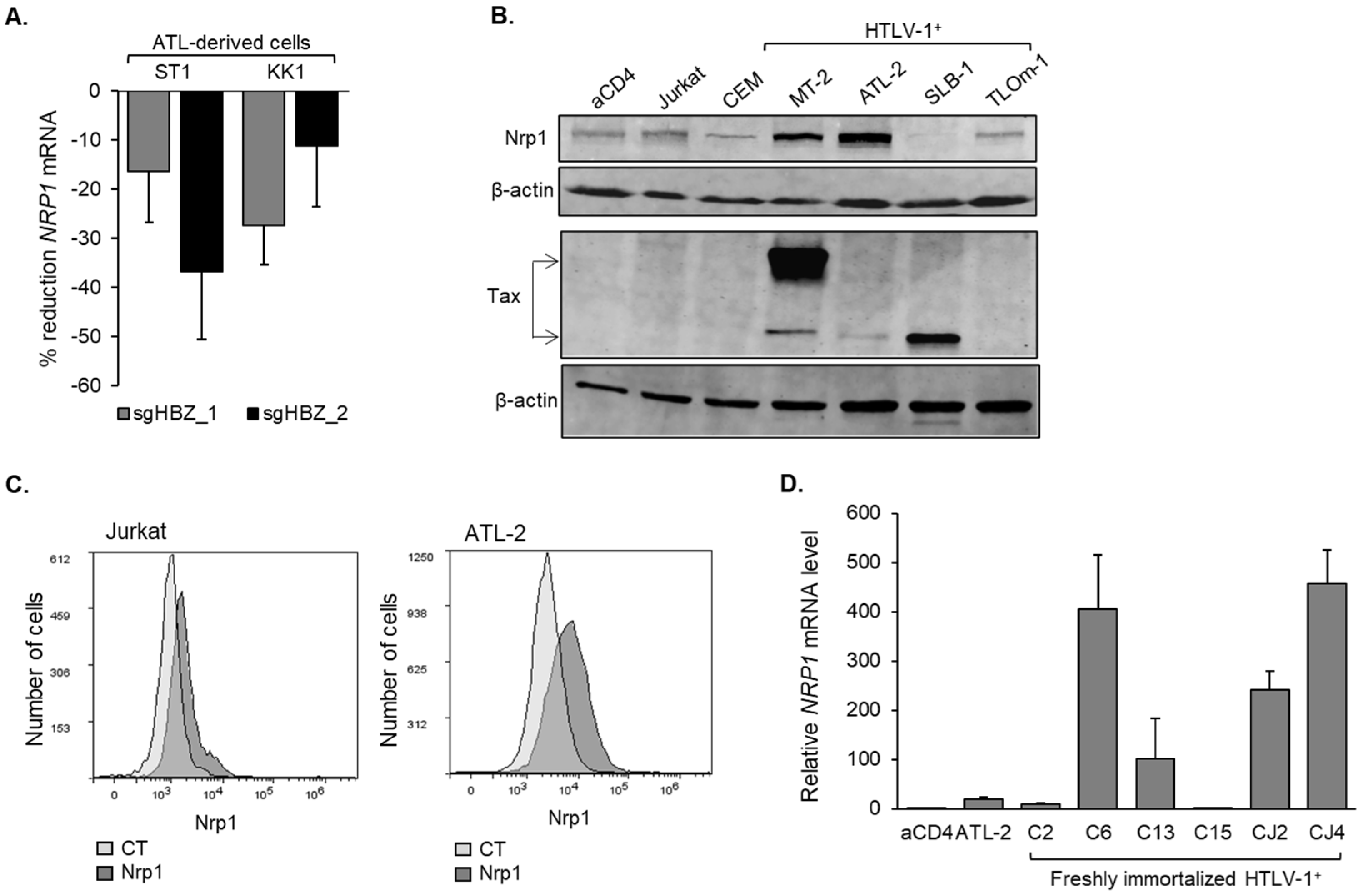
3.2. Nrp1 Expression Is Elevated in HTLV-1-Infected T-Cells Lines and Primary Cells Infected with HTLV-1
3.3. HBZ Activates NRP1 Transcription from an Enhancer Downstream of the Gene
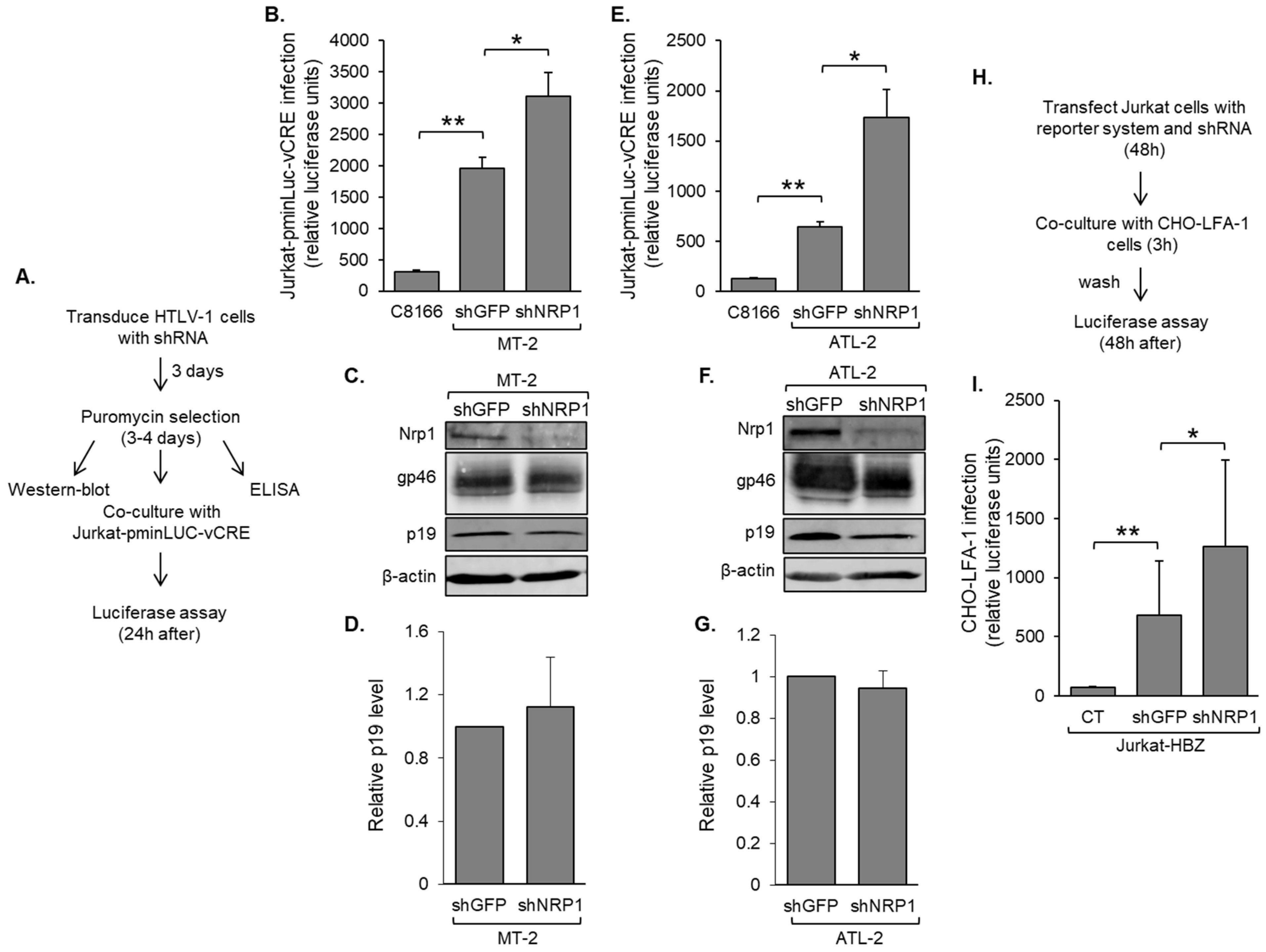
3.4. Nrp1 Expression in HTLV-1-Infected T-Cells Inhibits HTLV-1 Infection
3.5. Nrp1 Is Incorporated into the Viral Particle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [Green Version]
- Bangham, C.R.M. Human T Cell Leukemia Virus Type 1: Persistence and Pathogenesis. Annu. Rev. Immunol. 2018, 36, 43–71. [Google Scholar] [CrossRef]
- Yasunaga, J.I. Viral, genetic, and immune factors in the oncogenesis of adult T-cell leukemia/lymphoma. Int. J. Hematol. 2023, 117, 504–511. [Google Scholar] [CrossRef]
- Bangham, C.R.; Araujo, A.; Yamano, Y.; Taylor, G.P. HTLV-1-associated myelopathy/tropical spastic paraparesis. Nat. Rev. Dis. Prim. 2015, 1, 15012. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Taylor, G.P.; Jacobson, S. Inflammatory manifestations of HTLV-1 and their therapeutic options. Expert Rev. Clin. Immunol. 2014, 10, 1531–1546. [Google Scholar] [CrossRef]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Thoma-Kress, A.K. Molecular Mechanisms of HTLV-1 Cell-to-Cell Transmission. Viruses 2016, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.L. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Pinon, J.D.; Klasse, P.J.; Jassal, S.R.; Welson, S.; Weber, J.; Brighty, D.W.; Sattentau, Q.J. Human T-cell leukemia virus type 1 envelope glycoprotein gp46 interacts with cell surface heparan sulfate proteoglycans. J. Virol. 2003, 77, 9922–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghez, D.; Lepelletier, Y.; Lambert, S.; Fourneau, J.M.; Blot, V.; Janvier, S.; Arnulf, B.; van Endert, P.M.; Heveker, N.; Pique, C.; et al. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J. Virol. 2006, 80, 6844–6854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.S.; Lambert, S.; Bouttier, M.; Benit, L.; Ruscetti, F.W.; Hermine, O.; Pique, C. Molecular aspects of HTLV-1 entry: Functional domains of the HTLV-1 surface subunit (SU) and their relationships to the entry receptors. Viruses 2011, 3, 794–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Tessier-Lavigne, M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 1997, 90, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodkin, A.L.; Levengood, D.V.; Rowe, E.G.; Tai, Y.T.; Giger, R.J.; Ginty, D.D. Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- West, D.C.; Rees, C.G.; Duchesne, L.; Patey, S.J.; Terry, C.J.; Turnbull, J.E.; Delehedde, M.; Heegaard, C.W.; Allain, F.; Vanpouille, C.; et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J. Biol. Chem. 2005, 280, 13457–13464. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, A.; Gotze, T.; Korc, M. Hepatocyte growth factor-mediated cell invasion in pancreatic cancer cells is dependent on neuropilin-1. Cancer Res. 2007, 67, 10309–10316. [Google Scholar] [CrossRef] [Green Version]
- Dhar, K.; Dhar, G.; Majumder, M.; Haque, I.; Mehta, S.; Van Veldhuizen, P.J.; Banerjee, S.K.; Banerjee, S. Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1. Mol. Cancer 2010, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Glinka, Y.; Prud’homme, G.J. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 2008, 84, 302–310. [Google Scholar] [CrossRef]
- Prud’homme, G.J.; Glinka, Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 2012, 3, 921–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelland, T.; Djordjevic, S. Crystal Structure of the Neuropilin-1 MAM Domain: Completing the Neuropilin-1 Ectodomain Picture. Structure 2016, 24, 2008–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerc, I.; Polakowski, N.; Andre-Arpin, C.; Cook, P.; Barbeau, B.; Mesnard, J.M.; Lemasson, I. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J. Biol. Chem. 2008, 283, 23903–23913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, P.R.; Polakowski, N.; Lemasson, I. HTLV-1 HBZ protein deregulates interactions between cellular factors and the KIX domain of p300/CBP. J. Mol. Biol. 2011, 409, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Gaudray, G.; Gachon, F.; Basbous, J.; Biard-Piechaczyk, M.; Devaux, C.; Mesnard, J.M. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J. Virol. 2002, 76, 12813–12822. [Google Scholar] [CrossRef] [Green Version]
- Basbous, J.; Arpin, C.; Gaudray, G.; Piechaczyk, M.; Devaux, C.; Mesnard, J.M. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J. Biol. Chem. 2003, 278, 43620–46327. [Google Scholar] [CrossRef] [Green Version]
- Thebault, S.; Basbous, J.; Hivin, P.; Devaux, C.; Mesnard, J.M. HBZ interacts with JunD and stimulates its transcriptional activity. FEBS Lett. 2004, 562, 165–170. [Google Scholar] [CrossRef]
- Lemasson, I.; Lewis, M.R.; Polakowski, N.; Hivin, P.; Cavanagh, M.H.; Thebault, S.; Barbeau, B.; Nyborg, J.K.; Mesnard, J.M. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J. Virol. 2007, 81, 1543–1553. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, T.; Mukai, R.; Nakahara, N.; Matsumoto, J.; Isono, O.; Kobayashi, Y.; Takahashi, S.; Shimotohno, K. HTLV-1 basic leucine-zipper factor, HBZ, interacts with MafB and suppresses transcription through a Maf recognition element. J. Cell. Biochem. 2010, 111, 187–194. [Google Scholar] [CrossRef]
- Hagiya, K.; Yasunaga, J.; Satou, Y.; Ohshima, K.; Matsuoka, M. ATF3, an HTLV-1 bZip factor binding protein, promotes proliferation of adult T-cell leukemia cells. Retrovirology 2011, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushing, A.W.; Rushing, B.; Hoang, K.; Sanders, S.V.; Peloponese, J.M., Jr.; Polakowski, N.; Lemasson, I. HTLV-1 basic leucine zipper factor protects cells from oxidative stress by upregulating expression of Heme Oxygenase I. PLoS Pathog. 2019, 15, e1007922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinke, A.W.; Grigoryan, G.; Keating, A.E. Identification of bZIP interaction partners of viral proteins HBZ, MEQ, BZLF1, and K-bZIP using coiled-coil arrays. Biochemistry 2010, 49, 1985–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Coutts, A.; Xu, L.; Yu, J.; Ohshima, K.; Matsuoka, M. HTLV-1 bZIP factor supports proliferation of adult T cell leukemia cells through suppression of C/EBPalpha signaling. Retrovirology 2013, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Yasunaga, J.; Matsuoka, M. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 2016, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Alasiri, A.; Abboud Guerr, J.; Hall, W.W.; Sheehy, N. Novel Interactions between the Human T-Cell Leukemia Virus Type 1 Antisense Protein HBZ and the SWI/SNF Chromatin Remodeling Family: Implications for Viral Life Cycle. J. Virol. 2019, 93, e00412-19. [Google Scholar] [CrossRef] [Green Version]
- Polakowski, N.; Sarker, M.A.K.; Hoang, K.; Boateng, G.; Rushing, A.W.; Kendle, W.; Pique, C.; Green, P.L.; Panfil, A.R.; Lemasson, I. HBZ upregulates myoferlin expression to facilitate HTLV-1 infection. PLoS Pathog. 2023, 19, e1011202. [Google Scholar] [CrossRef]
- Fazio, A.L.; Kendle, W.; Hoang, K.; Korleski, E.; Lemasson, I.; Polakowski, N. Human T-Cell Leukemia Virus Type 1 (HTLV-1) bZIP Factor Upregulates the Expression of ICAM-1 To Facilitate HTLV-1 Infection. J. Virol. 2019, 93, e00608-19. [Google Scholar] [CrossRef]
- Barnard, A.L.; Igakura, T.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 2005, 106, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Millen, S.; Gross, C.; Donhauser, N.; Mann, M.C.; Peloponese, J.M., Jr.; Thoma-Kress, A.K. Collagen IV (COL4A1, COL4A2), a Component of the Viral Biofilm, Is Induced by the HTLV-1 Oncoprotein Tax and Impacts Virus Transmission. Front. Microbiol. 2019, 10, 2439. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, S.A.; Turpin, J.; Cachat, A.; Afonso, P.V.; Gessain, A.; Brady, J.N.; Pise-Masison, C.A.; Mahieux, R. Gem-induced cytoskeleton remodeling increases cellular migration of HTLV-1-infected cells, formation of infected-to-target T-cell conjugates and viral transmission. PLoS Pathog. 2014, 10, e1003917. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43 Pt A, 99–112. [Google Scholar]
- Rousset, R.; Desbois, C.; Bantignies, F.; Jalinot, P. Effects on NF-kappa B1/p105 processing of the interaction between the HTLV-1 transactivator Tax and the proteasome. Nature 1996, 381, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Laverdure, S.; Polakowski, N.; Hoang, K.; Lemasson, I. Permissive sense and antisense transcription from the 5’ and 3’ long terminal repeats of Human T-cell Leukemia Virus type 1. J. Virol. 2016, 90, 3600–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.C.; Kopp, F.; et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Maksimova, V.; Smith, S.; Seth, J.; Phelps, C.; Niewiesk, S.; Satou, Y.; Green, P.L.; Panfil, A.R. HTLV-1 intragenic viral enhancer influences immortalization phenotype in vitro, but is dispensable for persistence and disease development in animal models. Front. Immunol. 2022, 13, 954077. [Google Scholar] [CrossRef]
- Polakowski, N.; Gregory, H.; Mesnard, J.M.; Lemasson, I. Expression of a protein involved in bone resorption, Dkk1, is activated by HTLV-1 bZIP factor through its activation domain. Retrovirology 2010, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Lemasson, I.; Nyborg, J.K. Human T-cell leukemia virus type I Tax repression of p73beta is mediated through competition for the C/H1 domain of CBP. J. Biol. Chem. 2001, 276, 15720–15727. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.R.; Schroeder, M.; Fernandez, P.; Taubert, S.; Amati, B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001, 15, 2069–2082. [Google Scholar] [CrossRef] [Green Version]
- Lemasson, I.; Polakowski, N.; Laybourn, P.J.; Nyborg, J.K. Transcription regulatory complexes bind the human T-cell leukemia virus 5’ and 3’ long terminal repeats to control gene expression. Mol. Cell. Biol. 2004, 24, 6117–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Zhang, M.; Wright, G.; Huang, D.W.; Xiao, W.; Powell, J.; Petrus, M.N.; Yang, Y.; et al. Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell 2018, 34, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, Y.; Yap, S.F.; Ravichandran, V.; Matsumura, F.; Ito, M.; Spinelli, B.; Kelly, K. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell. Biol. 2002, 157, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.; Bouttier, M.; Vassy, R.; Seigneuret, M.; Petrow-Sadowski, C.; Janvier, S.; Heveker, N.; Ruscetti, F.W.; Perret, G.; Jones, K.S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. [Google Scholar] [CrossRef] [Green Version]
- Polakowski, N.; Lemasson, I. Regulation of HTLV-1 Transcription by Viral and Cellular Proteins. In Recent Advances in Human Retroviruses: Principles of Replication and Pathogenesis. Advances in Retroviral Research; Lever, A.M.L., Jeang, K.T., Eds.; World Scientific Publishing Co.: Singapore, 2010; pp. 129–169. [Google Scholar]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013, 41, D56–D63. [Google Scholar] [CrossRef]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Bhat, N.K.; Adachi, Y.; Samuel, K.P.; Derse, D. HTLV-1 gene expression by defective proviruses in an infected T-cell line. Virology 1993, 196, 15–24. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, L.; Zhang, X.; Zhang, J.; Shang, H.; Liang, G. Neuropilin-1, a myeloid cell-specific protein, is an inhibitor of HIV-1 infectivity. Proc. Natl. Acad. Sci. USA 2022, 119, e2114884119. [Google Scholar] [CrossRef]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef] [Green Version]
- Gillet, N.A.; Cook, L.; Laydon, D.J.; Hlela, C.; Verdonck, K.; Alvarez, C.; Gotuzzo, E.; Clark, D.; Farre, L.; Bittencourt, A.; et al. Strongyloidiasis and infective dermatitis alter human T lymphotropic virus-1 clonality in vivo. PLoS Pathog. 2013, 9, e1003263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.B.; Melamed, A.; Demontis, M.A.; Laydon, D.J.; Fox, J.M.; Tosswill, J.H.; de Freitas, D.; Price, A.D.; Medcalf, J.F.; Martin, F.; et al. Rapid dissemination of human T-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology 2016, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laydon, D.J.; Sunkara, V.; Boelen, L.; Bangham, C.R.M.; Asquith, B. The relative contributions of infectious and mitotic spread to HTLV-1 persistence. PLoS Comput. Biol. 2020, 16, e1007470. [Google Scholar] [CrossRef] [PubMed]
- Yasuma, K.; Yasunaga, J.; Takemoto, K.; Sugata, K.; Mitobe, Y.; Takenouchi, N.; Nakagawa, M.; Suzuki, Y.; Matsuoka, M. HTLV-1 bZIP Factor Impairs Anti-viral Immunity by Inducing Co-inhibitory Molecule, T Cell Immunoglobulin and ITIM Domain (TIGIT). PLoS Pathog. 2016, 12, e1005372. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Vinson, C.; Acharya, A.; Taparowsky, E.J. Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim. Biophys. Acta 2006, 1759, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Polakowski, N.; Pearce, M.; Kuguyo, O.; Boateng, G.; Hoang, K.; Lemasson, I. The splice 1 variant of HTLV-1 bZIP factor stabilizes c-Jun. Virology 2020, 549, 51–58. [Google Scholar] [CrossRef]
- Murata, K.; Hayashibara, T.; Sugahara, K.; Uemura, A.; Yamaguchi, T.; Harasawa, H.; Hasegawa, H.; Tsuruda, K.; Okazaki, T.; Koji, T.; et al. A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J. Virol. 2006, 80, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Yanagihara, K.; Tsukasaki, K.; Murata, K.; Hasegawa, H.; Yamada, Y.; Kamihira, S. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 2008, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Satou, Y.; Yasunaga, J.; Fujisawa, J.; Matsuoka, M. Transcriptional control of spliced and unspliced human T-cell leukemia virus type 1 bZIP factor (HBZ) gene. J. Virol. 2008, 82, 9359–9368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caceres, C.J.; Angulo, J.; Lowy, F.; Contreras, N.; Walters, B.; Olivares, E.; Allouche, D.; Merviel, A.; Pino, K.; Sargueil, B.; et al. Non-canonical translation initiation of the spliced mRNA encoding the human T-cell leukemia virus type 1 basic leucine zipper protein. Nucleic Acids Res. 2018, 46, 11030–11047. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.S.; Villaudy, J.; Gazzolo, L.; Castellazzi, M.; Mesnard, J.M.; Duc Dodon, M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 2007, 4, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Satou, Y.; Sugata, K.; Miyazato, P.; Green, P.L.; Imamura, T.; Matsuoka, M. HTLV-1 bZIP factor enhances TGF-beta signaling through p300 coactivator. Blood 2011, 118, 1865–1876. [Google Scholar] [CrossRef] [Green Version]
- Polakowski, N.; Terol, M.; Hoang, K.; Nash, I.; Laverdure, S.; Gazon, H.; Belrose, G.; Mesnard, J.M.; Cesaire, R.; Peloponese, J.M.; et al. HBZ stimulates brain-derived neurotrophic factor/TrkB autocrine/paracrine signaling to promote survival of human T-cell leukemia virus type 1-Infected T cells. J. Virol. 2014, 88, 13482–13494. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, M.; Meier, C.; Mann, A.M.; Chapman, N.; Wasiak, A. Envelope glycoprotein of HIV induces interference and cytolysis resistance in CD4+ cells: Mechanism for persistence in AIDS. Cell 1988, 53, 483–496. [Google Scholar] [CrossRef]
- Weller, S.K.; Joy, A.E.; Temin, H.M. Correlation between cell killing and massive second-round superinfection by members of some subgroups of avian leukosis virus. J. Virol. 1980, 33, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Lindwasser, O.W.; Chaudhuri, R.; Bonifacino, J.S. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr. Mol. Med. 2007, 7, 171–184. [Google Scholar] [CrossRef]
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13, 735. [Google Scholar] [CrossRef]
- Al-Saleem, J.; Dirksen, W.P.; Martinez, M.P.; Shkriabai, N.; Kvaratskhelia, M.; Ratner, L.; Green, P.L. HTLV-1 Tax-1 interacts with SNX27 to regulate cellular localization of the HTLV-1 receptor molecule, GLUT1. PLoS ONE 2019, 14, e0214059. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.B.; Melamed, A.; Niederer, H.; Valganon, M.; Laydon, D.; Foroni, L.; Taylor, G.P.; Matsuoka, M.; Bangham, C.R. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood 2014, 123, 3925–3931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederer, H.A.; Laydon, D.J.; Melamed, A.; Elemans, M.; Asquith, B.; Matsuoka, M.; Bangham, C.R. HTLV-1 proviral integration sites differ between asymptomatic carriers and patients with HAM/TSP. Virol. J. 2014, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, Y.; Yasunaga, J.I.; Matsuoka, M. HTLV-1’s Foxy Strategy for Survival and Transmission. Front. Virol. 2022, 1, 792659. [Google Scholar] [CrossRef]
- Powell, J.; Mota, F.; Steadman, D.; Soudy, C.; Miyauchi, J.T.; Crosby, S.; Jarvis, A.; Reisinger, T.; Winfield, N.; Evans, G.; et al. Small Molecule Neuropilin-1 Antagonists Combine Antiangiogenic and Antitumor Activity with Immune Modulation through Reduction of Transforming Growth Factor Beta (TGFbeta) Production in Regulatory T-Cells. J. Med. Chem. 2018, 61, 4135–4154. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kendle, W.; Hoang, K.; Korleski, E.; Panfil, A.R.; Polakowski, N.; Lemasson, I. Upregulation of Neuropilin-1 Inhibits HTLV-1 Infection. Pathogens 2023, 12, 831. https://doi.org/10.3390/pathogens12060831
Kendle W, Hoang K, Korleski E, Panfil AR, Polakowski N, Lemasson I. Upregulation of Neuropilin-1 Inhibits HTLV-1 Infection. Pathogens. 2023; 12(6):831. https://doi.org/10.3390/pathogens12060831
Chicago/Turabian StyleKendle, Wesley, Kimson Hoang, Erica Korleski, Amanda R. Panfil, Nicholas Polakowski, and Isabelle Lemasson. 2023. "Upregulation of Neuropilin-1 Inhibits HTLV-1 Infection" Pathogens 12, no. 6: 831. https://doi.org/10.3390/pathogens12060831
APA StyleKendle, W., Hoang, K., Korleski, E., Panfil, A. R., Polakowski, N., & Lemasson, I. (2023). Upregulation of Neuropilin-1 Inhibits HTLV-1 Infection. Pathogens, 12(6), 831. https://doi.org/10.3390/pathogens12060831