
Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms

 , , , and
, , , and 
Abstract
:1. Introduction
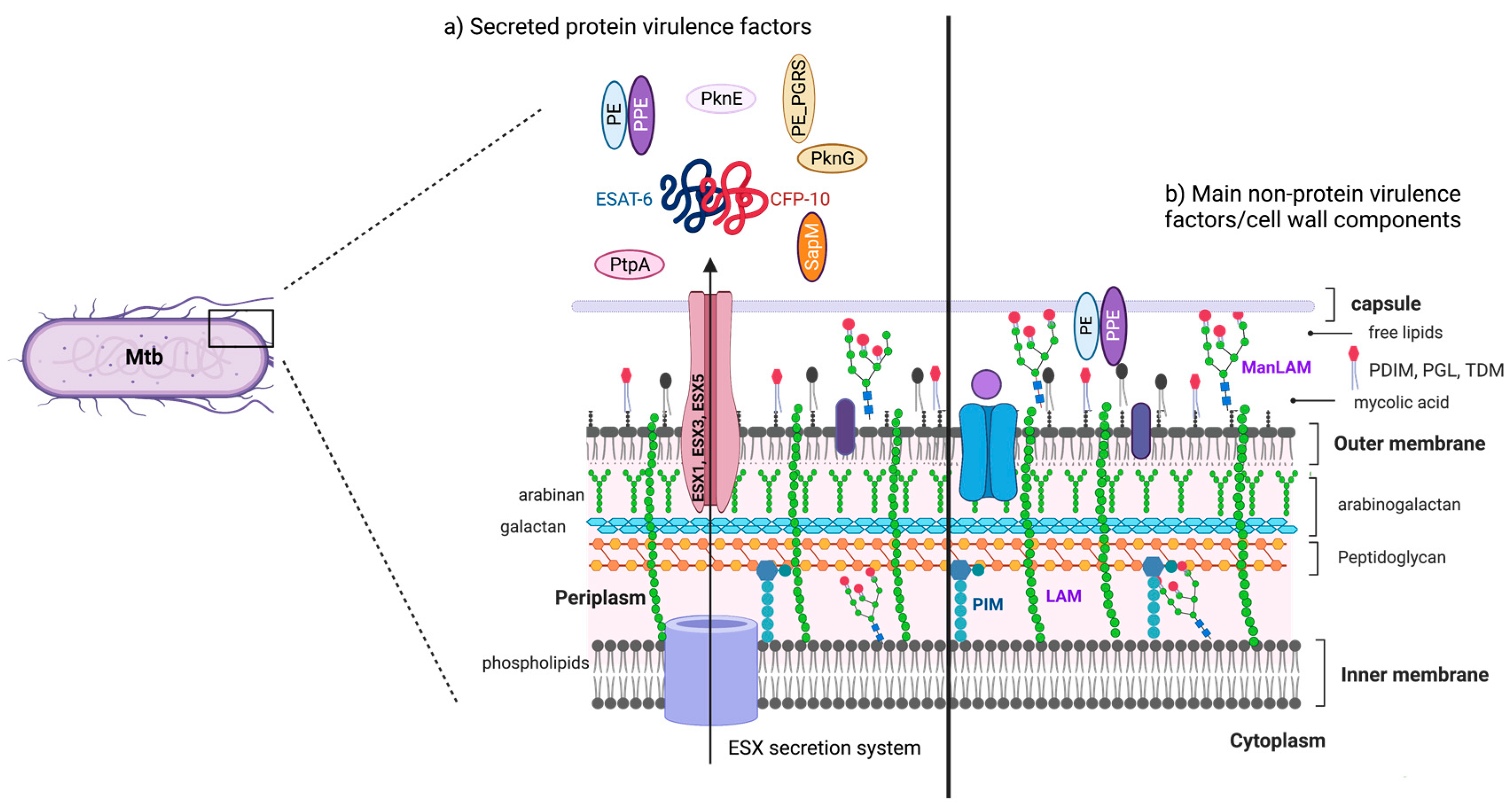
2. Mtb Virulence Factors: Who Are Involved in Disturbing Cell Death?
2.1. Non-Protein Virulence Factors
Major Non-Protein Virulence Factors Inside the Cell Wall
- Phosphatidyl-myo-inositol mannosides (PIMs) are the most abundant glycolipids in the mycobacterial cell envelope and are precursors of lipomannan (LM) and lipoarabinomannan (LAM) [17]. The PIMs comprise variable numbers of mannose units and levels of acylation. Virulent species possess PIMs with five or six mannoses that bind to the mannose receptor (MR), contributing to macrophage uptake. A few of the mannoses also interact with the dendritic cell (DC)-specific intercellular adhesion molecule-3-grabbing non-integrin DC-SIGN from DC [2,18]. Acyl phosphatidyl-myo-inositol dimannoside (AcPIM2) is a part of the inner membrane in most mycobacterial species, and AcPIM6 is involved in maintaining cell envelope integrity [16];
- LM is a multi-glycosylated lipid or polymannosylated PIMs, which is the basic structure of LAM. LM efficiently activates the innate immune response via a tetra-acylated form that activates macrophages through toll-like receptor-2 and 4 (TLR2 and TLR4, respectively). In contrast, its di-acylated form regulates and inhibits nitric oxide (NO) production and cytokine secretion in activated macrophages [19];
- LAM is a glycolipoconjugate composed of LM bound to multiple arabinose residues. When LAM acquires extra and random formation of mannose-capped LM, it is called ManLAM. An addition of phosphoinositol-capped LM makes it known as PILAM, and uncapped or arabinofuranosyl-terminated LM makes it AraLAM. PIMs, LM, LAM, and ManLAM, are recognized by specific receptors, expressed on the cell surface of antigen-presenting cells, such as MR, DC-SIGN, and dendritic cell activating receptor (DCAR), facilitating Mtb uptake into host cells [20,21]. Indeed, the diversity in Mtb mannosylated cell walls alters its virulence, affecting pathogeny and host adaptation [21];
- Phthiocerol dimycocerosates (PDIM) are lipids present on the outer leaflet of the outer membrane that may be secreted or shed from mycobacteria. PDIM promotes Mtb’s evasion of TLR-mediated pathogen detection, delaying the recruitment of immune cells and adaptive responses to the infection site [22]. In addition, PDIM has been suggested to be crucial for phagocytosis and in the rupture of phagosomes and mitochondrial membranes [22,23].
2.2. Protein Virulence Factors
2.2.1. Major Proteins Virulence Factors from Mtb
2.2.2. Major Lipoproteins Virulence Factors from Mtb
2.2.3. Major Phosphatases and Kinases Virulence Factors from Mtb
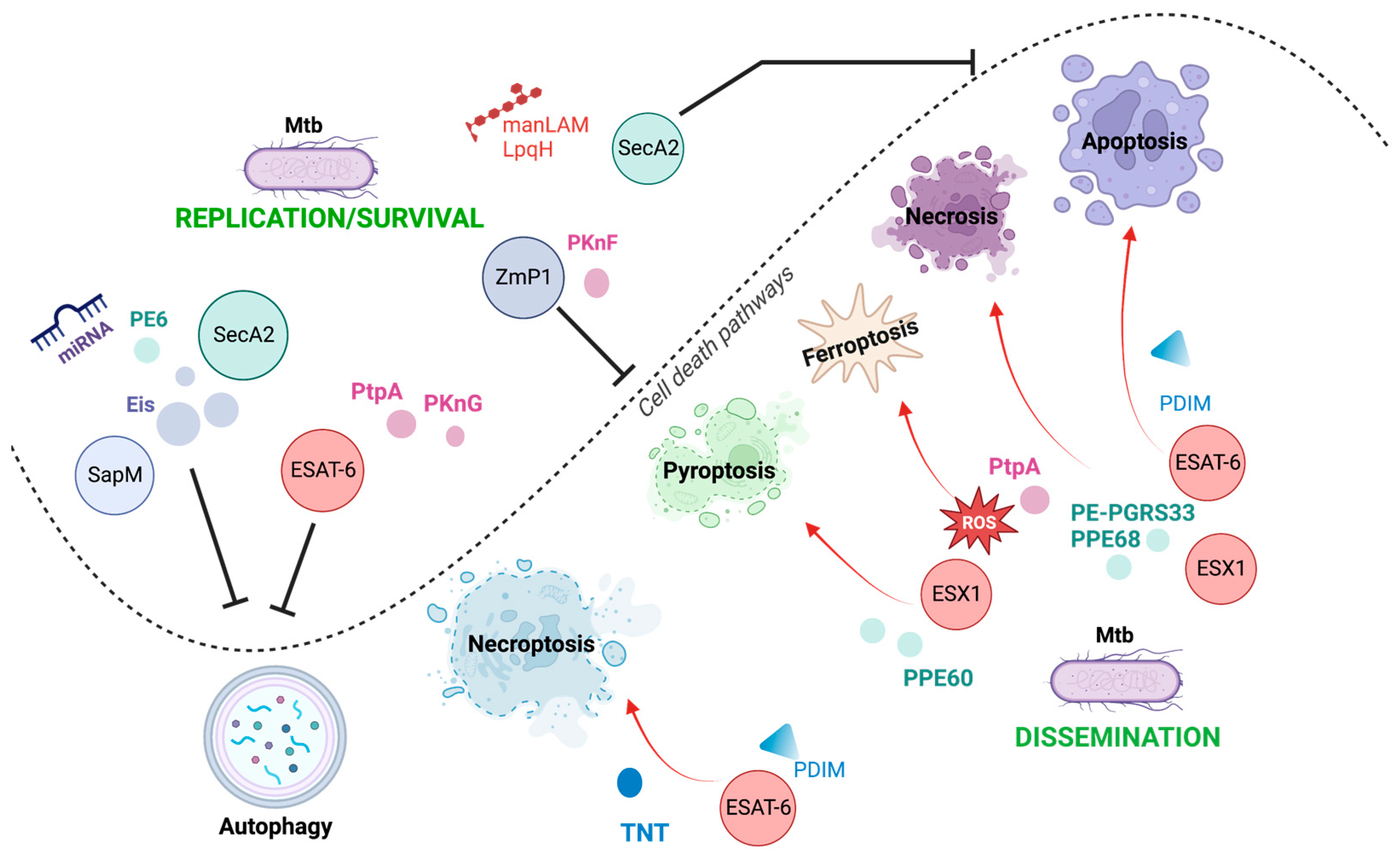
3. Cell Death Mechanisms Activated by Mtb Virulence Factors: The Good and the Bad
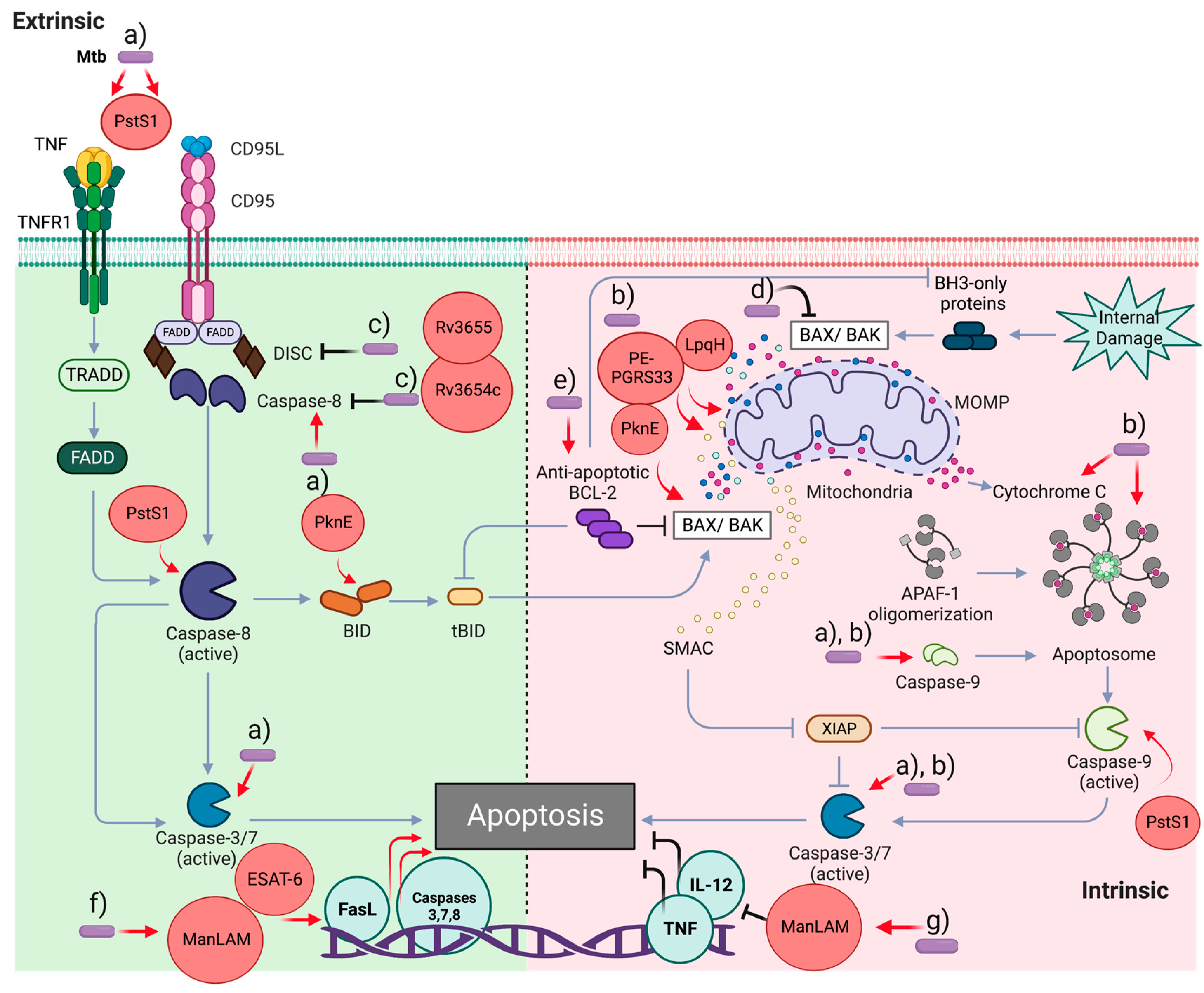
3.1. Apoptosis
3.1.1. Modulation of Apoptosis in the Mtb Infection: Proteins Involved
3.1.2. Modulation of Apoptosis in the Mtb Infection: Lipid Molecules Involved
3.2. Necrosis
Necrosis Modulation in Mycobacterial Infection
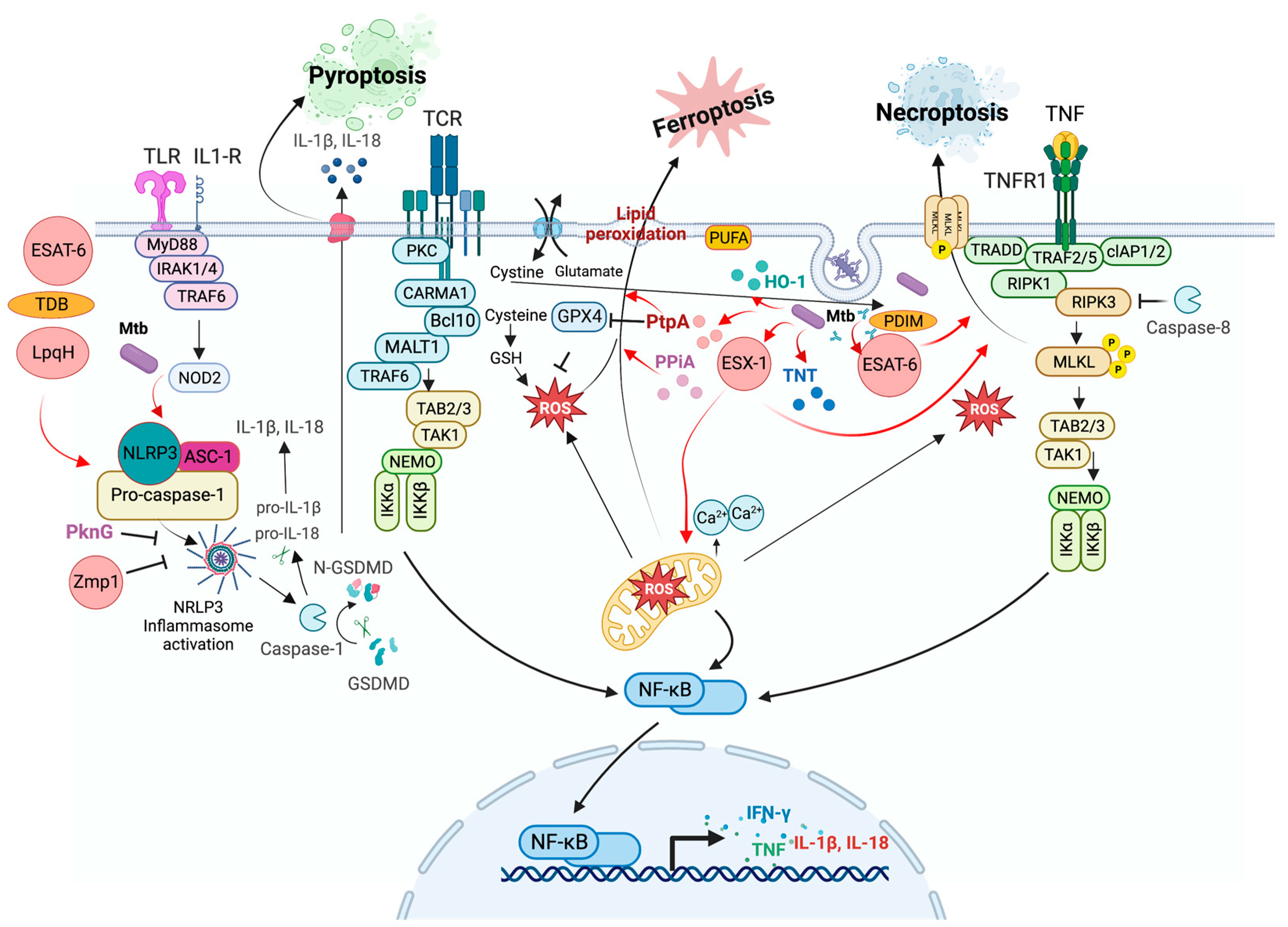
3.3. Pyroptosis
Modulation of Pyroptosis by Mtb
3.4. Necroptosis
Modulation of Necroptosis by Mtb
3.5. Autophagy-Dependent Cell Death
Modulation of Autophagy by Mtb
3.6. Ferroptosis
Modulation of Ferroptosis by Mtb
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 16076. [Google Scholar] [CrossRef]
- Echeverria-Valencia, G.; Flores-Villalva, S.; Espitia, C.I. Virulence Factors and Pathogenicity of Mycobacterium; Ribón, W., Ed.; IntechOpen: Rijeka, Croatia, 2017; p. Ch. 12. ISBN 978-1-78923-211-0. [Google Scholar]
- Global Tuberculosis Report 2022. Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 2 February 2023).
- Lee, J.; Boyce, S.; Powers, J.; Baer, C.; Sassetti, C.M.; Behar, S.M. CD11cHi Monocyte-Derived Macrophages Are a Major Cellular Compartment Infected by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008621. [Google Scholar] [CrossRef] [PubMed]
- Nadolinskaia, N.I.; Kotliarova, M.S.; Goncharenko, A.V. Fighting Tuberculosis: In Search of a BCG Replacement. Microorganisms 2022, 11, 51. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohareer, K.; Asalla, S.; Banerjee, S. Cell Death at the cross Roads of Host-Pathogen Interaction in Mycobacterium tuberculosis Infection. Tuberculosis 2018, 113, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492. [Google Scholar] [CrossRef]
- Feng, Z.; Bai, X.; Wang, T.; Garcia, C.; Bai, A.; Li, L.; Honda, J.R.; Nie, X.; Chan, E.D. Differential Responses by Human Macrophages to Infection With Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2020, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.E.; Brodin, P.; Jang, J.; Jang, M.S.; Robertson, B.D.; Gicquel, B.; Stewart, G.R. The Balance of Apoptotic and Necrotic Cell Death in Mycobacterium tuberculosis Infected Macrophages Is Not Dependent on Bacterial Virulence. PLoS ONE 2012, 7, e47573. [Google Scholar] [CrossRef] [Green Version]
- Coscolla, M.; Gagneux, S. Consequences of Genomic Diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzheimer, M.; Buter, J.; Minnaard, A.J. Chemical Synthesis of Cell Wall Constituents of Mycobacterium tuberculosis. Chem. Rev. 2021, 121, 9554–9643. [Google Scholar] [CrossRef]
- Chiaradia, L.; Lefebvre, C.; Parra, J.; Marcoux, J.; Burlet-Schiltz, O.; Etienne, G.; Tropis, M.; Daffé, M. Dissecting the Mycobacterial Cell Envelope and Defining the Composition of the Native Mycomembrane. Sci. Rep. 2017, 7, 12807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and Virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef] [PubMed]
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Fine Discrimination in the Recognition of Individual Species of Phosphatidyl-Myo-Inositol Mannosides from Mycobacterium tuberculosis by C-Type Lectin Pattern Recognition Receptors. J. Immunol. 2006, 177, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Doz, E.; Rose, S.; Nigou, J.; Gilleron, M.; Puzo, G.; Erard, F.; Ryffel, B.; Quesniaux, V.F.J. Acylation Determines the Toll-like Receptor (TLR)-Dependent Positive versus TLR2-, Mannose Receptor-, and SIGNR1-Independent Negative Regulation of pro-Inflammatory Cytokines by Mycobacterial Lipomannan. J. Biol. Chem. 2007, 282, 26014–26025. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.; Torrelles, J.B. Mannose-Capped Lipoarabinomannan in Mycobacterium tuberculosis Pathogenesis. Pathog. Dis. 2018, 76, fty026. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Rens, C.; Chao, J.D.; Sexton, D.L.; Tocheva, E.I.; Av-Gay, Y. Roles for Phthiocerol Dimycocerosate Lipids in Mycobacterium tuberculosis Pathogenesis. Microbiology 2021, 167, 001042. [Google Scholar] [CrossRef]
- Day, T.A.; Mittler, J.E.; Nixon, M.R.; Thompson, C.; Miner, M.D.; Hickey, M.J.; Liao, R.P.; Pang, J.M.; Shayakhmetov, D.M.; Sherman, D.R. Mycobacterium tuberculosis Strains Lacking Surface Lipid Phthiocerol Dimycocerosate Are Susceptible to Killing by an Early Innate Host Response. Infect. Immun. 2014, 82, 5214–5222. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Ghatak, D.; Das, P.; BoseDasgupta, S. ESX Secretion System: The Gatekeepers of Mycobacterial Survivability and Pathogenesis. Eur. J. Microbiol. Immunol. 2020, 10, 202–209. [Google Scholar] [CrossRef]
- Dumas, E.; Boritsch, E.C.; Vandenbogaert, M.; De La Vega, R.C.R.; Thiberge, J.M.; Caro, V.; Gaillard, J.L.; Heym, B.; Girard-Misguich, F.; Brosch, R.; et al. Mycobacterial Pan-Genome Analysis Suggests Important Role of Plasmids in the Radiation of Type VII Secretion Systems. Genome Biol. Evol. 2016, 8, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Briken, V. Modular Organization of the ESX-5 Secretion System in Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2016, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augenstreich, J.; Briken, V. Host Cell Targets of Released Lipid and Secreted Protein Effectors of Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2020, 10, 595029. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-W. The Role of ESX-1 in Mycobacterium tuberculosis Pathogenesis. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Peng, X.; Sun, J. Mechanism of ESAT-6 Membrane Interaction and Its Roles in Pathogenesis of Mycobacterium tuberculosis. Toxicon 2016, 116, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cheng, H.F.; Zhou, J.; Chan, C.Y.; Lau, K.F.; Tsui, S.K.W.; Au, S.W. ngor Structural Basis of the PE–PPE Protein Interaction in Mycobacterium tuberculosis. J. Biol. Chem. 2017, 292, 16880. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Pathak, S.K.; Banerjee, A.; Pathak, S.; Bhattacharyya, A.; Yang, Z.; Talarico, S.; Kundu, M.; Basu, J. Execution of Macrophage Apoptosis by PE_PGRS33 of Mycobacterium tuberculosis Is Mediated by Toll-like Receptor 2-Dependent Release of Tumor Necrosis Factor-Alpha. J. Biol. Chem. 2007, 282, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucci, I.; Camassa, S.; Cascioferro, A.; Sali, M.; Anoosheh, S.; Zumbo, A.; Minerva, M.; Iantomasi, R.; De Maio, F.; Di Sante, G.; et al. PE-PGRS33 Contributes to Mycobacterium tuberculosis Entry in Macrophages through Interaction with TLR2. PLoS ONE 2016, 11, e0150800. [Google Scholar] [CrossRef]
- Shukla, S.; Richardson, E.T.; Drage, M.G.; Boom, W.H.; Harding, C.V. Mycobacterium tuberculosis Lipoprotein and Lipoglycan Binding to Toll-like Receptor 2 Correlates with Agonist Activity and Functional Outcomes. Infect. Immunol. 2018, 86, e00450-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, A.J.; Dobos, K.M.; Belisle, J.T.; Harding, C.V.; Boom, W.H. Mycobacterium tuberculosis LprG (Rv1411c): A Novel TLR-2 Ligand That Inhibits Human Macrophage Class II MHC Antigen Processing. J. Immunol. 2004, 173, 2660–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Richardson, E.T.; Athman, J.J.; Shi, L.; Wearsch, P.A.; McDonald, D.; Banaei, N.; Boom, W.H.; Jackson, M.; Harding, C.V. Mycobacterium tuberculosis Lipoprotein LprG Binds Lipoarabinomannan and Determines Its Cell Envelope Localization to Control Phagolysosomal Fusion. PLoS Pathog. 2014, 10, e1004471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, K.; Sinha, A.Y.; Ghorpade, D.S.; Togarsimalemath, S.K.; Patil, S.A.; Kaveri, S.V.; Balaji, K.N.; Bayry, J. Src Homology 3-Interacting Domain of Rv1917c of Mycobacterium tuberculosis Induces Selective Maturation of Human Dendritic Cells by Regulating PI3K-MAPK-NF-KappaB Signaling and Drives Th2 Immune Responses. J. Biol. Chem. 2010, 285, 36511–36522. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Zhu, S.; Zhu, L.; Huang, W.; Wang, H.; Zhang, Z.; Xu, Y. Recombinant Lipoprotein Rv1016c Derived from Mycobacterium tuberculosis Is a TLR-2 Ligand That Induces Macrophages Apoptosis and Inhibits MHC II Antigen Processing. Front. Cell Infect. Microbiol. 2016, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Esparza, M.; Palomares, B.; García, T.; Espinosa, P.; Zenteno, E.; Mancilla, R. PstS-1, the 38-KDa Mycobacterium tuberculosis Glycoprotein, Is an Adhesin, Which Binds the Macrophage Mannose Receptor and Promotes Phagocytosis. Scand. J. Immunol. 2015, 81, 46–55. [Google Scholar] [CrossRef]
- Saleh, M.T.; Belisle, J.T. Secretion of an Acid Phosphatase (SapM) by Mycobacterium tuberculosis That Is Similar to Eukaryotic Acid Phosphatases. J. Bacteriol. 2000, 182, 6850–6853. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.V.; Reddy, P.V.; Tyagi, A.K. Secreted Acid Phosphatase (SapM) of Mycobacterium tuberculosis Is Indispensable for Arresting Phagosomal Maturation and Growth of the Pathogen in Guinea Pig Tissues. PLoS ONE 2013, 8, e70514. [Google Scholar] [CrossRef] [Green Version]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis Virulence Is Mediated by PtpA Dephosphorylation of Human Vacuolar Protein Sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ge, P.; Qiang, L.; Tian, F.; Zhao, D.; Chai, Q.; Zhu, M.; Zhou, R.; Meng, G.; Iwakura, Y.; et al. The Mycobacterial Phosphatase PtpA Regulates the Expression of Host Genes and Promotes Cell Proliferation. Nat. Commun. 2017 8:1 2017, 8, 244. [Google Scholar] [CrossRef]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis Protein Tyrosine Phosphatase (PtpA) Excludes Host Vacuolar-H+-ATPase to Inhibit Phagosome Acidification. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Narayanan, S. PknE, a Serine/Threonine Kinase of Mycobacterium tuberculosis Modulates Multiple Apoptotic Paradigms. Infect. Genet. Evol. 2012, 12, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Lei, Z.; Yu, Y.; Lu, Z.; Qiang, L.; Chai, Q.; Zhang, Y.; Zhao, D.; Li, B.; Pang, Y.; et al. M. Tuberculosis PknG Manipulates Host Autophagy Flux to Promote Pathogen Intracellular Survival. Autophagy 2022, 18, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Ellinwood, S.; Augenstreich, J.; Mayer-Barber, K.D.; Briken, V. Mycobacterium tuberculosis Inhibits the NLRP3 Inflammasome Activation via Its Phosphokinase PknF. PLoS Pathog. 2021, 17, e1009712. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; Hasnain, S.E.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-like Receptor 4. mBio 2018, 9, e01017-18. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium tuberculosis. Cell. Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef]
- Lossi, L. The Concept of Intrinsic versus Extrinsic Apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef]
- Negulescu, A.M.; Mehlen, P. Dependence Receptors—The Dark Side Awakens. FEBS J. 2018, 285, 3909–3924. [Google Scholar] [CrossRef] [Green Version]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, Function, and Biotechnological Aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Li, X.; Li, F.; Zhang, X.; Zhang, H.; Zhao, Q.; Li, M.; Wu, X.; Wang, L.; Liu, J.; Wu, X.; et al. Caspase-8 Auto-Cleavage Regulates Programmed Cell Death and Collaborates with RIPK3/MLKL to Prevent Lymphopenia. Cell Death Differ. 2022, 29, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic Insights into Caspase-9 Activation by the Structure of the Apoptosome Holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein TBID Can Act as a BAX-like Effector of Apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Wallace, R.J. Clinical and Taxonomic Status of Pathogenic Nonpigmented or Late-Pigmenting Rapidly Growing Mycobacteria. Clin. Microbiol. Rev. 2002, 15, 716–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadifar, S.; Mostafaei, S.; Behrouzi, A.; Fateh, A.; Riahi, P.; Siadat, S.D.; Vaziri, F. Strain-Specific Behavior of Mycobacterium tuberculosis in A549 Lung Cancer Cell Line. BMC Bioinform. 2021, 22, 154. [Google Scholar] [CrossRef]
- Alves da Silva, D.A.; Da Silva, M.V.; Oliveira Barros, C.C.; Dias Alexandre, P.B.; Timóteo, R.P.; Catarino, J.S.; Sales-Campos, H.; Machado, J.R.; Rodrigues, D.B.R.; Oliveira, C.J.; et al. TNF-α Blockade Impairs in Vitro Tuberculous Granuloma Formation and down Modulate Th1, Th17 and Treg Cytokines. PLoS ONE 2018, 13, e0194430. [Google Scholar] [CrossRef] [Green Version]
- Arbués, A.; Brees, D.; Chibout, S.D.; Fox, T.; Kammüller, M.; Portevin, D. TNF-α Antagonists Differentially Induce TGF-Β1-Dependent Resuscitation of Dormant-like Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008312. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, J.; Dai, F.; Zhang, D.; Li, W. DUSP1 Mediates BCG Induced Apoptosis and Inflammatory Response in THP-1 Cells via MAPKs/NF-ΚB Signaling Pathway. Sci. Rep. 2023, 13, 2606. [Google Scholar] [CrossRef]
- Riendeau, C.J.; Kornfeld, H. THP-1 Cell Apoptosis in Response to Mycobacterial Infection. Infect. Immunol. 2003, 71, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.F.; Alves, C.C.S.; Figueiredo, B.B.M.; Rezende, A.B.; Wohlres-Viana, S.; da Silva, V.L.; Machado, M.A.; Teixeira, H.C. Tumour Necrosis Factor Receptors and Apoptosis of Alveolar Macrophages during Early Infection with Attenuated and Virulent Mycobacterium Bovis. Immunology 2013, 139, 503–512. [Google Scholar] [CrossRef]
- Woo, M.; Wood, C.; Kwon, D.; Park, K.H.P.; Fejer, G.; Delorme, V. Mycobacterium tuberculosis Infection and Innate Responses in a New Model of Lung Alveolar Macrophages. Front. Immunol. 2018, 9, 438. [Google Scholar] [CrossRef]
- Bohsali, A.; Abdalla, H.; Velmurugan, K.; Briken, V. The Non-Pathogenic Mycobacteria M. Smegmatis and M. Fortuitum Induce Rapid Host Cell Apoptosis via a Caspase-3 and TNF Dependent Pathway. BMC Microbiol. 2010, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Walters, A.; Keeton, R.; Labuschagné, A.; Hsu, N.J.; Jacobs, M. TNFRp75-Dependent Immune Regulation of Alveolar Macrophages and Neutrophils during Early Mycobacterium tuberculosis and Mycobacterium Bovis BCG Infection. Immunology 2021, 162, 220–234. [Google Scholar] [CrossRef]
- Balcewicz-Sablinska, M.K.; Keane, J.; Kornfeld, H.; Remold, H.G. Pathogenic Mycobacterium tuberculosis Evades Apoptosis of Host Macrophages by Release of TNF-R2, Resulting in Inactivation of TNF-α. J. Immunol. 1998, 161, 2636–2641. [Google Scholar] [CrossRef]
- Kundu, M.; Pathak, S.K.; Kumawat, K.; Basu, S.; Chatterjee, G.; Pathak, S.; Noguchi, T.; Takeda, K.; Ichijo, H.; Thien, C.B.F.; et al. A TNF- and c-Cbl-Dependent FLIP(S)-Degradation Pathway and Its Function in Mycobacterium tuberculosis-Induced Macrophage Apoptosis. Nat. Immunol. 2009, 10, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Danelishvili, L.; Yamazaki, Y.; Selker, J.; Bermudez, L.E. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c Proteins Participate in the Suppression of Macrophage Apoptosis. PLoS ONE 2010, 5, e10474. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramarska, E.; Squeglia, F.; De Maio, F.; Delogu, G.; Berisio, R. PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells 2021, 10, 161. [Google Scholar] [CrossRef]
- Augenstreich, J.; Arbues, A.; Simeone, R.; Haanappel, E.; Wegener, A.; Sayes, F.; Le Chevalier, F.; Chalut, C.; Malaga, W.; Guilhot, C.; et al. ESX-1 and Phthiocerol Dimycocerosates of Mycobacterium tuberculosis Act in Concert to Cause Phagosomal Rupture and Host Cell Apoptosis. Cell Microbiol. 2017, 19, e12726. [Google Scholar] [CrossRef] [Green Version]
- Derrick, S.C.; Morris, S.L. The ESAT6 Protein of Mycobacterium tuberculosis Induces Apoptosis of Macrophages by Activating Caspase Expression. Cell Microbiol. 2007, 9, 1547–1555. [Google Scholar] [CrossRef]
- Francis, R.J.; Butler, R.E.; Stewart, G.R. Mycobacterium tuberculosis ESAT-6 Is a Leukocidin Causing Ca2+ Influx, Necrosis and Neutrophil Extracellular Trap Formation. Cell Death Dis. 2014, 5, e1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Li, F.; Jia, S.; Zhang, K.; Jiang, W.; Shang, Y.; Chang, K.; Deng, S.; Chen, M. Early Secreted Antigen ESAT-6 of Mycobacterium tuberculosis Promotes Apoptosis of Macrophages via Targeting the MicroRNA155-SOCS1 Interaction. Cell Physiol. Biochem. 2015, 35, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Velmurugan, K.; Cowan, M.J.; Briken, V. The Type I NADH Dehydrogenase of Mycobacterium tuberculosis Counters Phagosomal NOX2 Activity to Inhibit TNF-Alpha-Mediated Host Cell Apoptosis. PLoS Pathog. 2010, 6, e1000864. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Nieuwenhuizen, N.; Vogelzang, A.; Liu, H.; Kaiser, P.; Schuerer, S.; Lazar, D.; Wagner, I.; Mollenkopf, H.J.; Kaufmann, S.H.E. Deletion of NuoG from the Vaccine Candidate Mycobacterium Bovis BCG ΔureC::Hly Improves Protection against Tuberculosis. mBio 2016, 7, e00679-16. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, M.; Bensing, B.A.; Sullam, P.M. The Two Distinct Types of SecA2-Dependent Export Systems. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Sullivan, J.T.; Young, E.F.; Mccann, J.R.; Braunstein, M. The Mycobacterium tuberculosis SecA2 System Subverts Phagosome Maturation to Promote Growth in Macrophages. Infect. Immunol. 2012, 80, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Hinchey, J.; Lee, S.; Jeon, B.Y.; Basaraba, R.J.; Venkataswamy, M.M.; Chen, B.; Chan, J.; Braunstein, M.; Orme, I.M.; Derrick, S.C.; et al. Enhanced Priming of Adaptive Immunity by a Proapoptotic Mutant of Mycobacterium tuberculosis. J. Clin. Investig. 2007, 117, 2279–2288. [Google Scholar] [CrossRef]
- Jayakumar, D.; Jacobs, W.R.; Narayanan, S. Protein Kinase E of Mycobacterium tuberculosis Has a Role in the Nitric Oxide Stress Response and Apoptosis in a Human Macrophage Model of Infection. Cell Microbiol. 2008, 10, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chen, Z.; Bai, N.; Yan, R.; Xu, M.; Wu, W.; Liang, W.; Li, H.; Mao, Y. Construction of a Eukaryotic Expression System with Stable and Secretory Expression of Mycobacterium tuberculosis 38 KDa Protein. World J. Microbiol. Biotechnol. 2021, 37, 175. [Google Scholar] [CrossRef]
- Sanchez, A.; Espinosa, P.; Esparza, M.A.; Colon, M.; Bernal, G.; Mancilla, R. Mycobacterium tuberculosis 38-KDa Lipoprotein Is Apoptogenic for Human Monocyte-Derived Macrophages. Scand. J. Immunol. 2009, 69, 20–28. [Google Scholar] [CrossRef]
- Wojtas, B.; Fijalkowska, B.; Wlodarczyk, A.; Schollenberger, A.; Niemialtowski, M.; Hamasur, B.; Pawlowski, A.; Krzyzowska, M. Mannosylated Lipoarabinomannan Balances Apoptosis and Inflammatory State in Mycobacteria-Infected and Uninfected Bystander Macrophages. Microb. Pathog. 2011, 51, 9–21. [Google Scholar] [CrossRef]
- Dao, D.N.; Kremer, L.; Guérardel, Y.; Molano, A.; Jacobs, W.R.; Porcelli, S.A.; Briken, V. Mycobacterium tuberculosis Lipomannan Induces Apoptosis and Interleukin-12 Production in Macrophages. Infect. Immun. 2004, 72, 2067–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, P.; Kumar, R.; Jana, K.; Chakraborty, S.; Ghosh, Z.; Kundu, M.; Basu, J. Gene Expression Profiling of Mycobacterium tuberculosis Lipoarabinomannan-Treated Macrophages: A Role of the Bcl-2 Family Member A1 in Inhibition of Apoptosis in Mycobacteria-Infected Macrophages. IUBMB Life 2015, 67, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Kundapura, S.V.; Basak, A.J.; Mukherjee, D.; Dash, S.; Ganguli, N.; Das, A.K.; Mukherjee, G.; Samanta, D.; Ramagopal, U.A. High-Resolution Crystal Structure of LpqH, an Immunomodulatory Surface Lipoprotein of Mycobacterium tuberculosis Reveals a Distinct Fold and a Conserved Cleft on Its Surface. Int. J. Biol. Macromol. 2022, 210, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Espinosa, P.; García, T.; Mancilla, R. The 19 KDa Mycobacterium tuberculosis Lipoprotein (LpqH) Induces Macrophage Apoptosis through Extrinsic and Intrinsic Pathways: A Role for the Mitochondrial Apoptosis-Inducing Factor. Clin. Dev. Immunol. 2012, 2012, 950503. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Payá, E.; Orzáez, M.; Mondragón, L.; Wolan, D.; Wells, J.A.; Messeguer, A.; Vicent, M.J. Molecules That Modulate Apaf-1 Activity. Med. Res. Rev. 2011, 31, 649–675. [Google Scholar] [CrossRef]
- Orzáez, M.; Gortat, A.; Mondragón, L.; Pérez-Payá, E. Peptides and Peptide Mimics as Modulators of Apoptotic Pathways. ChemMedChem 2009, 4, 146–160. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as Anticancer Mechanism: Function and Dysfunction of Its Modulators and Targeted Therapeutic Strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Aging 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis Evades Macrophage Defenses by Inhibiting Plasma Membrane Repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.G.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis Replicates within Necrotic Human Macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Mahamed, D.; Boulle, M.; Ganga, Y.; Mc Arthur, C.; Skroch, S.; Oom, L.; Catinas, O.; Pillay, K.; Naicker, M.; Rampersad, S.; et al. Intracellular Growth of Mycobacterium tuberculosis after Macrophage Cell Death Leads to Serial Killing of Host Cells. Elife 2017, 6, e22028. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E.M. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.W.; Jacobs, W.R. Mycobacterium tuberculosis Exploits Human Interferon γ to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J. Infect. Dis. 2013, 208, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, H.; Li, H.; Dang, G.; Cui, Z.; Song, N.; Wang, Q.; Liu, S.; Chen, L. PE17 Protein from Mycobacterium tuberculosis Enhances Mycobacterium Smegmatis Survival in Macrophages and Pathogenicity in Mice. Microb. Pathog. 2019, 126, 63–73. [Google Scholar] [CrossRef]
- Peng, X.; Luo, T.; Zhai, X.; Zhang, C.; Suo, J.; Ma, P.; Wang, C.; Bao, L. PPE11 of Mycobacterium tuberculosis Can Alter Host Inflammatory Response and Trigger Cell Death. Microb. Pathog. 2019, 126, 45–55. [Google Scholar] [CrossRef]
- Danelishvili, L.; Everman, J.; Bermudez, L.E. Mycobacterium tuberculosis PPE68 and Rv2626c Genes Contribute to the Host Cell Necrosis and Bacterial Escape from Macrophages. Virulence 2016, 7, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Riaz, S.M.; Bjune, G.A.; Wiker, H.G.; Sviland, L.; Mustafa, T. Mycobacterial Antigens Accumulation in Foamy Macrophages in Murine Pulmonary Tuberculosis Lesions: Association with Necrosis and Making of Cavities. Scand. J. Immunol. 2020, 91, e12866. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Repasy, T.; Papavinasasundaram, K.; Sassetti, C.; Kornfeld, H. Mycobacterium tuberculosis Induces an Atypical Cell Death Mode to Escape from Infected Macrophages. PLoS ONE 2011, 6, e18367. [Google Scholar] [CrossRef] [PubMed]
- Afriyie-Asante, A.; Dabla, A.; Dagenais, A.; Berton, S.; Smyth, R.; Sun, J. Mycobacterium tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front. Immunol. 2021, 12, 4401. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, S.; Briken, V. Interaction of Mycobacteria with Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, J.; Zhao, Z.; Xin, T.; Fan, X.; Shen, Q.; Raheem, A.; Lee, C.R.; Jiang, H.; Ding, J. Regulated Necrosis, a Proinflammatory Cell Death, Potentially Counteracts Pathogenic Infections. Cell Death Dis. 2022, 13, 637. [Google Scholar] [CrossRef]
- Eklund, D.; Welin, A.; Andersson, H.; Verma, D.; Söderkvist, P.; Stendahl, O.; Särndahl, E.; Lerm, M. Human Gene Variants Linked to Enhanced NLRP3 Activity Limit Intramacrophage Growth of Mycobacterium tuberculosis. J. Infect. Dis. 2014, 209, 749. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis during Mycobacterium tuberculosis Infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Moura-Alves, P.; Sonawane, A.; Hacohen, N.; Griffiths, G.; Moita, L.F.; Anes, E. Mycobacterium tuberculosis Protein ESAT-6 Is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol. 2010, 12, 1046–1063. [Google Scholar] [CrossRef]
- Gong, Z.; Kuang, Z.; Li, H.; Li, C.; Ali, M.K.; Huang, F.; Li, P.; Li, Q.; Huang, X.; Ren, S.; et al. Regulation of Host Cell Pyroptosis and Cytokines Production by Mycobacterium tuberculosis Effector PPE60 Requires LUBAC Mediated NF-ΚB Signaling. Cell Immunol. 2019, 335, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Hu, Y.; Zhang, X.; Chi, M.; Xu, S.; Wang, H.; Zhang, X. Mycobacterium tuberculosis PE_PGRS19 Induces Pyroptosis through a Non-Classical Caspase-11/GSDMD Pathway in Macrophages. Microorganisms 2022, 10, 2473. [Google Scholar] [CrossRef]
- Kurane, T.; Matsunaga, T.; Ida, T.; Sawada, K.; Nishimura, A.; Fukui, M.; Umemura, M.; Nakayama, M.; Ohara, N.; Matsumoto, S.; et al. GRIM-19 Is a Target of Mycobacterial Zn2+ Metalloprotease 1 and Indispensable for NLRP3 Inflammasome Activation. FASEB J. 2022, 36, e22096. [Google Scholar] [CrossRef]
- Danelishvili, L.; Everman, J.L.; McNamara, M.J.; Bermudez, L.E. Inhibition of the Plasma-Membrane-Associated Serine Protease Cathepsin G by Mycobacterium tuberculosis Rv3364c Suppresses Caspase-1 and Pyroptosis in Macrophages. Front. Microbiol. 2012, 2, 281. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in Development and Diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef]
- Chirieleison, S.M.; Kertesy, S.B.; Abbott, D.W. Synthetic Biology Reveals the Uniqueness of the RIP Kinase Domain. J. Immunol. 2016, 196, 4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, E.; Park, K.A.; Shen, H.M.; Hur, G.M. The Resurrection of RIP Kinase 1 as an Early Cell Death Checkpoint Regulator-a Potential Target for Therapy in the Necroptosis Era. Exp. Mol. Med. 2022, 54, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, L.; Jia, J.; Tian, W.; Lai, R.; Zhang, Z.; Li, J.; Ju, J.; Xu, H. Knowledge Mapping of Necroptosis From 2012 to 2021: A Bibliometric Analysis. Front. Immunol. 2022, 13, 2930. [Google Scholar] [CrossRef] [PubMed]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic Signaling Is Primed in Mycobacterium tuberculosis-Infected Macrophages, but Its Pathophysiological Consequence in Disease Is Restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; He, Y.; Jiang, Y.; Wu, Q.; Liu, Y.; Xie, Q.; Zou, Y.; Wu, J.; Zhang, C.; Zhou, Z.; et al. PRMT1 Reverts the Immune Escape of Necroptotic Colon Cancer through RIP3 Methylation. Cell Death Dis. 2023, 14, 233. [Google Scholar] [CrossRef]
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA Amplifies Necroptosis Signaling by Activating Cytosolic DNA Sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL Forms Cation Channels. Cell Res. 2016, 26, 517. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of Cell-Surface Proteases Promotes Necroptosis, Inflammation and Cell Migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP Controls Necroptosis through Ubiquitylation- and Lysosome-Dependent Degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Molnár, T.; Pallagi, P.; Tél, B.; Király, R.; Csoma, E.; Jenei, V.; Varga, Z.; Gogolák, P.; Odile Hueber, A.; Máté, Z.; et al. Caspase-9 Acts as a Regulator of Necroptotic Cell Death. FEBS J. 2021, 288, 6476–6491. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Ramakrishnan, L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD+ Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429. [Google Scholar] [CrossRef] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Niederweis, M. NAD Hydrolysis by the Tuberculosis Necrotizing Toxin Induces Lethal Oxidative Stress in Macrophages. Cell Microbiol. 2020, 22, e13115. [Google Scholar] [CrossRef]
- Quigley, J.; Hughitt, V.K.; Velikovsky, C.A.; Mariuzza, R.A.; El-Sayed, N.M.; Briken, V. The Cell Wall Lipid PDIM Contributes to Phagosomal Escape and Host Cell Exit of Mycobacterium tuberculosis. mBio 2017, 8, e00148-17. [Google Scholar] [CrossRef] [Green Version]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 Maintains Epithelial Homeostasis by Inhibiting Apoptosis and Necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Yanti, B.; Mulyadi, M.; Amin, M.; Harapan, H.; Mertaniasih, N.M.; Soetjipto, S. The Role of Mycobacterium tuberculosis Complex Species on Apoptosis and Necroptosis State of Macrophages Derived from Active Pulmonary Tuberculosis Patients. BMC Res. Notes 2020, 13, 415. [Google Scholar] [CrossRef]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol.-Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Sanicas, M.; Nigou, J.; Guilhot, C.; Astarie-Dequeker, C.; Vergne, I. The Lipid Virulence Factors of Mycobacterium tuberculosis Exert Multilayered Control over Autophagy-Related Pathways in Infected Human Macrophages. Cells 2020, 9, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Kim, J.K.; Jung, C.H.; Kim, Y.J.; Jung, E.J.; Lee, S.H.; Choi, H.R.; Son, Y.S.; Shim, S.M.; Jeon, S.M.; et al. Chemical Modulation of SQSTM1/P62-Mediated Xenophagy That Targets a Broad Range of Pathogenic Bacteria. Autophagy 2022, 18, 2926–2945. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Jeon, B.-Y.; Lee, H.-M.; Jin, H.S.; Yuk, J.-M.; Song, C.-H.; Lee, S.-H.; Lee, Z.-W.; Cho, S.-N.; Kim, J.-M.; et al. Mycobacterium tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death through Redox-Dependent Signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [Green Version]
- Pahari, S.; Negi, S.; Aqdas, M.; Arnett, E.; Schlesinger, L.S.; Agrewala, J.N. Induction of Autophagy through CLEC4E in Combination with TLR4: An Innovative Strategy to Restrict the Survival of Mycobacterium tuberculosis. Autophagy 2020, 16, 1021–1043. [Google Scholar] [CrossRef]
- Sharma, N.; Shariq, M.; Quadir, N.; Singh, J.; Sheikh, J.A.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis Protein PE6 (Rv0335c), a Novel TLR4 Agonist, Evokes an Inflammatory Response and Modulates the Cell Death Pathways in Macrophages to Enhance Intracellular Survival. Front. Immunol. 2021, 12, 696491. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, R.; Zhang, R.; Wu, J.; Hu, D.; Xing, Y.; Ni, S.; Tie, B. [ESAT-6 inhibits autophagy in macrophages and promotes the growth of BCG]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2017, 33, 310–314. [Google Scholar]
- Zulauf, K.E.; Sullivan, J.T.; Braunstein, M. The SecA2 Pathway of Mycobacterium tuberculosis Exports Effectors That Work in Concert to Arrest Phagosome and Autophagosome Maturation. PLoS Pathog. 2018, 14, e1007011. [Google Scholar] [CrossRef]
- Vergne, I.; Gilleron, M.; Nigou, J. Manipulation of the Endocytic Pathway and Phagocyte Functions by Mycobacterium tuberculosis Lipoarabinomannan. Front. Cell Infect. Microbiol. 2014, 4, 187. [Google Scholar] [CrossRef] [Green Version]
- Amaral, E.P.; Foreman, T.W.; Namasivayam, S.; Hilligan, K.L.; Kauffman, K.D.; Bomfim, C.C.B.; Costa, D.L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; Santana, M.F.; et al. GPX4 Regulates Cellular Necrosis and Host Resistance in Mycobacterium tuberculosis Infection. J. Exp. Med. 2022, 219, e20220504. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ouyang, J.; Lu, Y.; Harypursat, V.; Chen, Y. A Dual Role of Heme Oxygenase-1 in Tuberculosis. Front. Immunol. 2022, 13, 842858. [Google Scholar] [CrossRef]
- Amaral, E.P.; Namasivayam, S.; Costa, D.L.; Fisher, L.; Bomfim, C.C.B.; Andrade, B.B.; Sher, A. The Transcription Factor BACH1 Promotes Tissue Damage and Host Susceptibility in Mycobacterium tuberculosis Infection by Reducing Expression of Gpx4, a Major Negative Regulator of Ferroptosis. J. Immunol. 2020, 204, 227.16. [Google Scholar] [CrossRef]
- Ma, C.; Wu, X.; Zhang, X.; Liu, X.; Deng, G. Corrigendum: Heme Oxygenase-1 Modulates Ferroptosis by Fine-Tuning Levels of Intracellular Iron and Reactive Oxygen Species of Macrophages in Response to Bacillus Calmette-Guerin Infection. Front. Cell Infect. Microbiol. 2022, 12, 1105336. [Google Scholar] [CrossRef]
- Fan, J.; Jiang, T.; He, D. Emerging Insights into the Role of Ferroptosis in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2023, 14, 1579. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhang, Y.; Lei, Z.; Lu, Z.; Tan, S.; Ge, P.; Chai, Q.; Zhao, M.; Zhang, X.; Li, B.; et al. A Mycobacterial Effector Promotes Ferroptosis-Dependent Pathogenicity and Dissemination. Nat. Commun. 2023, 14, 1430. [Google Scholar] [CrossRef]
- Dubey, N.; Khan, M.Z.; Kumar, S.; Sharma, A.; Das, L.; Bhaduri, A.; Singh, Y.; Nandicoori, V.K. Mycobacterium tuberculosis Peptidyl Prolyl Isomerase A Interacts With Host Integrin Receptor to Exacerbate Disease Progression. J. Infect. Dis. 2021, 224, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Neyrolles, O. Die Another Way: Ferroptosis Drives Tuberculosis Pathology. J Exp Med 2019, 216, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tang, P.; Cao, J.; Song, Q.; Zhu, L.; Ma, S.; Zhang, J. Lipid Peroxidation Aggravates Anti-Tuberculosis Drug-Induced Liver Injury: Evidence of Ferroptosis Induction. Biochem. Biophys. Res. Commun. 2020, 533, 1512–1518. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J.F. Ferroptosis: A Mixed Blessing for Infectious Diseases. Front. Pharmacol. 2022, 13, 992734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, Y.; Hu, L.; Fu, J.; Li, D.; Chen, J.; Long, Y. Inhibition of HSPA8 by Rifampicin Contributes to Ferroptosis via Enhancing Autophagy. Liver Int. 2022, 42, 2889–2899. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Chen, J.; Xu, G.Y.; Zhang, Z.; Xue, J.; Zeng, H.; Jiang, J.; Chen, T.; Qin, Z.; Li, H.; et al. Ferroptosis-Related Gene SOCS1, a Marker for Tuberculosis Diagnosis and Treatment, Involves in Macrophage Polarization and Facilitates Bone Destruction in Tuberculosis. Tuberculosis 2022, 132, 102140. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mtb Virulence Factor/Protein or Lipid Origin | Abbreviation | Function/Impact on Cell Death | Cell Type | Dissemination/ Intracellular Survival | References |
|---|---|---|---|---|---|
| Dual-Specificity Phosphatase 1 | DUSP1 | Proapoptotic, a regulator of MAPKs/NF-κB signaling pathway, induces apoptosis | THP-1 macrophages | Undefined | [62] |
| Rv3654c and Rv3655c putative proteins | Antiapoptotic interfering with the extrinsic pathway | Human monocytic cell line U937 | Undefined | [70] | |
| PE family proteins | PE-PGRS33, PE_PGRS5 (Rv0297) | Proapoptotic and induces necrosis | RAW 264.7 murine macrophages | Undefined | [72] |
| PE family proteins | PE17 (Rv1646) | Inhibits IL-6, IL-12, and TNF production and induces macrophage necrosis. | Mouse peritoneal macrophages, THP-1 macrophages, J774A.1 mouse cells, and BMDM C57BL/6 | ||
| PPE11 (Rv0453) | Induces macrophage necrosis | Intracellular survival | [102,103,104] | ||
| PPE68-Rv2626c | Induces necrosis | [119,120] | |||
| PPE60, and PE_PGRS19 | Induces pyroptosis by a non-classical-pathway | Dissemination | [149,150] | ||
| PE6 | Suppresses Autophagy | ||||
| Early secretory antigenic target 6 kD | ESAT-6 (EsxA) | Proapoptotic, increases gene expression of caspases-3, -5, -7, and -8. Promotes inflammasome activation-pyroptosis induction. Induces neutrophil necrosis and NETs production, and loss of integrity of the plasma membrane and triggers necroptosis Inhibits phagosome maturation | hMDM and THP-1 macrophages | [73,74,75] | |
| [116,117,118] | |||||
| Dissemination | [28,29] | ||||
| Zmp1, and Rv3364c | Inhibit inflammasome activation and pyroptosis | [121,122] | |||
| Subunit of type I NADH dehydrogenase | nuoG | Antiapoptotic, inhibits apoptosis in the infected cells | THP-1 macrophages, BMDM BALB/c mice | Undefined | [77,78] |
| ATPase of the Sec secretion system | SecA2 | Antiapoptotic, secretion of several proteins involved in blocking apoptosis | Undefined | [79] | |
| Serine threonine protein kinases | PknE | Proapoptotic | THP-1 macrophages | Intracellular survival | [44,82] |
| PknF | NLRP3 inflammasome inhibition | BMDM | [46] | ||
| Protein kinase G | PknG | Phagosome arresting, inhibition of phagosome–lysosome fusion | J774 macrophage cells | Intracellular survival | [45] |
| Phosphatase | SapM | Phagosome arresting, inhibition of phagosome–lysosome fusion | RAW 264.7 murine macrophages, THP-1 macrophages, and guinea pig tissues | Intracellular survival | [39,40] |
| Protein tyrosine phosphatase | PtpA | Phagosome arresting, inhibition phagosome acidification | THP-1 macrophages | Intracellular survival | [41,42,43] |
| Triggers ferroptosis | Dissemination | [159] | |||
| 38 kDa lipoprotein | PstS1 | Proapoptotic, TLR-2 and caspases -8, -9, and -3 activation, and promotes phagocytosis | hMDM | [84] | |
| Tuberculosis necrotizing toxin | TNT | Degrades all intracellular NAD+ and triggers necroptosis | THP-1 macrophages | Undefined | [137,138] |
| Enhanced intracellular survival protein | Eis | Abrogates ROS and proinflammatory cytokines production. Inhibits autophagosome formation | THP-1 macrophages | Undefined | [9,148] |
| Phthiocerol dimycocerosates | PDIMs | Proapoptotic, increases gene expression of caspases-3, -5, -7, and -8 | hMDM | Undefined | [73] |
| Induces necroptosis | THP-1 macrophages | [139] | |||
| Lipomannan (Glycolipid) | LM | Proapoptotic | THP-1 macrophages | Undefined | [86] |
| Mannosylated lipoarabinomannan (glycolipid) | ManLAM | Antiapoptotic, up-regulates Bcl-2 depending on the virulence | THP-1 macrophages, BMDM BALB/c mice | Undefined | [85,87] |
| Lipoprotein | LpqH | Proapoptotic, promotes apoptosome formation and activation of procaspases -9 and -3 | hMDM | Undefined | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramon-Luing, L.A.; Palacios, Y.; Ruiz, A.; Téllez-Navarrete, N.A.; Chavez-Galan, L. Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens 2023, 12, 839. https://doi.org/10.3390/pathogens12060839
Ramon-Luing LA, Palacios Y, Ruiz A, Téllez-Navarrete NA, Chavez-Galan L. Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens. 2023; 12(6):839. https://doi.org/10.3390/pathogens12060839
Chicago/Turabian StyleRamon-Luing, Lucero A., Yadira Palacios, Andy Ruiz, Norma A. Téllez-Navarrete, and Leslie Chavez-Galan. 2023. "Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms" Pathogens 12, no. 6: 839. https://doi.org/10.3390/pathogens12060839
APA StyleRamon-Luing, L. A., Palacios, Y., Ruiz, A., Téllez-Navarrete, N. A., & Chavez-Galan, L. (2023). Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens, 12(6), 839. https://doi.org/10.3390/pathogens12060839