
Role of TAM Receptors in Antimalarial Humoral Immune Response


{kind=link}
{kind=link}
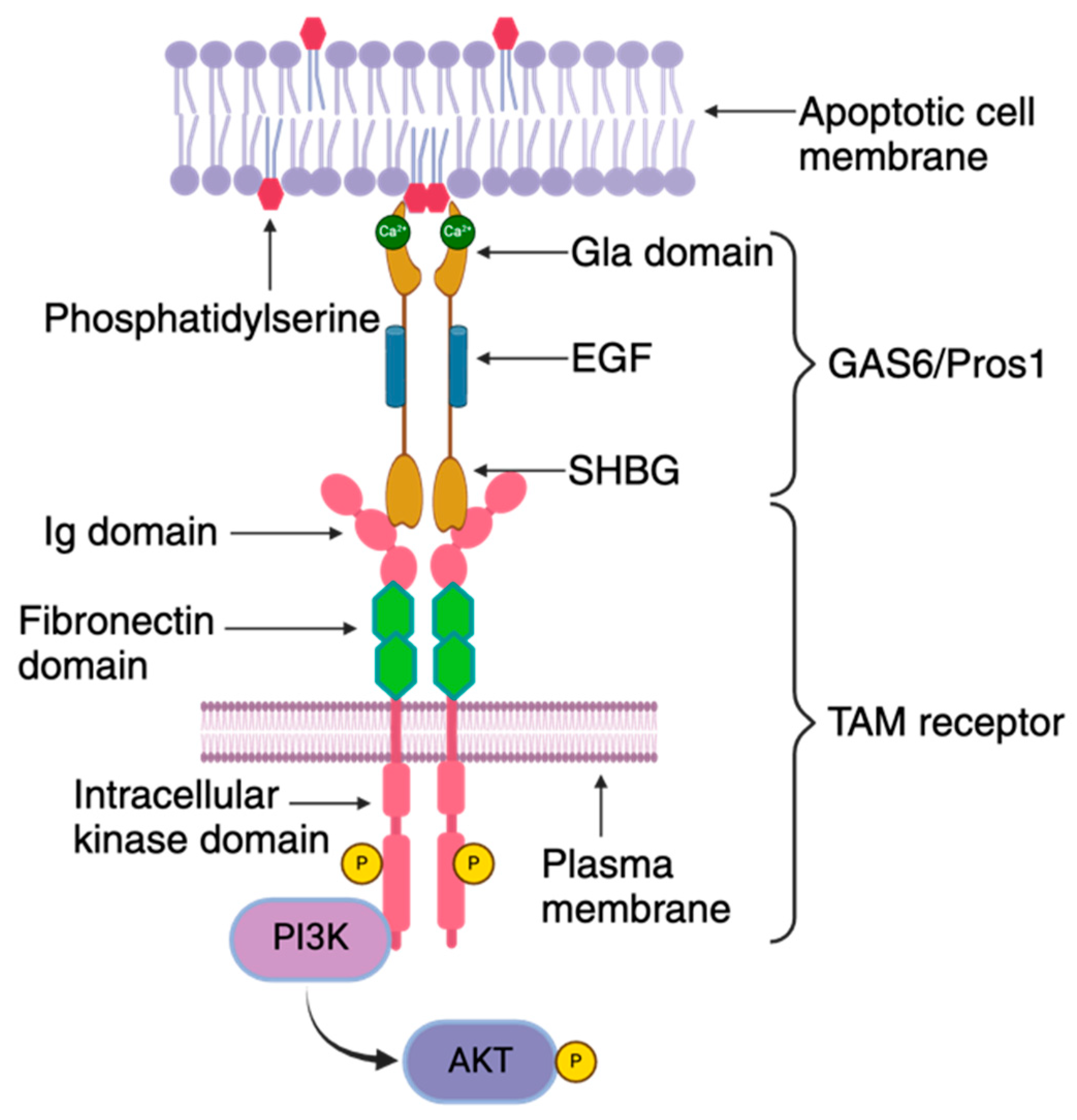{kind=link}
{kind=link}
Abstract
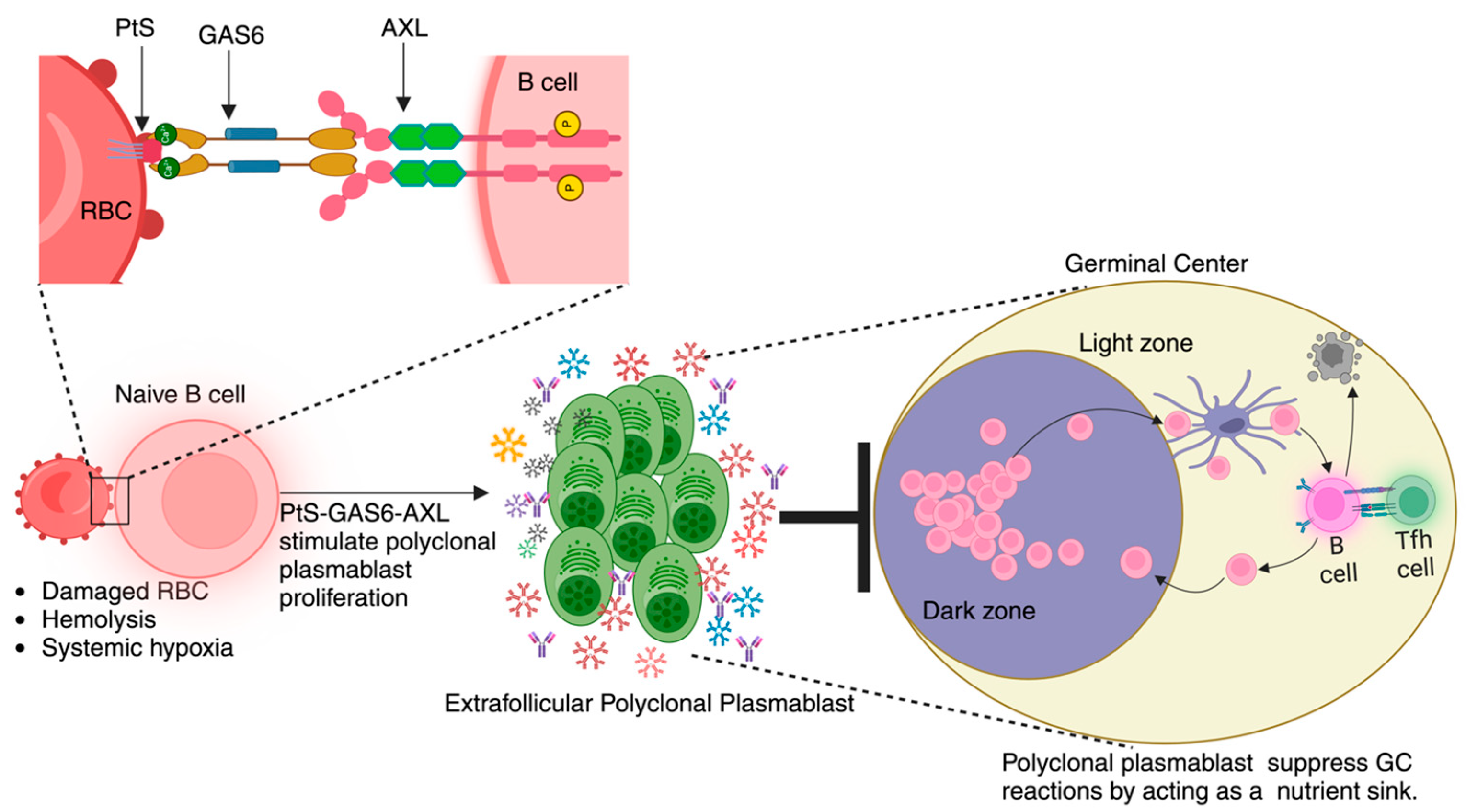
:1. Introduction
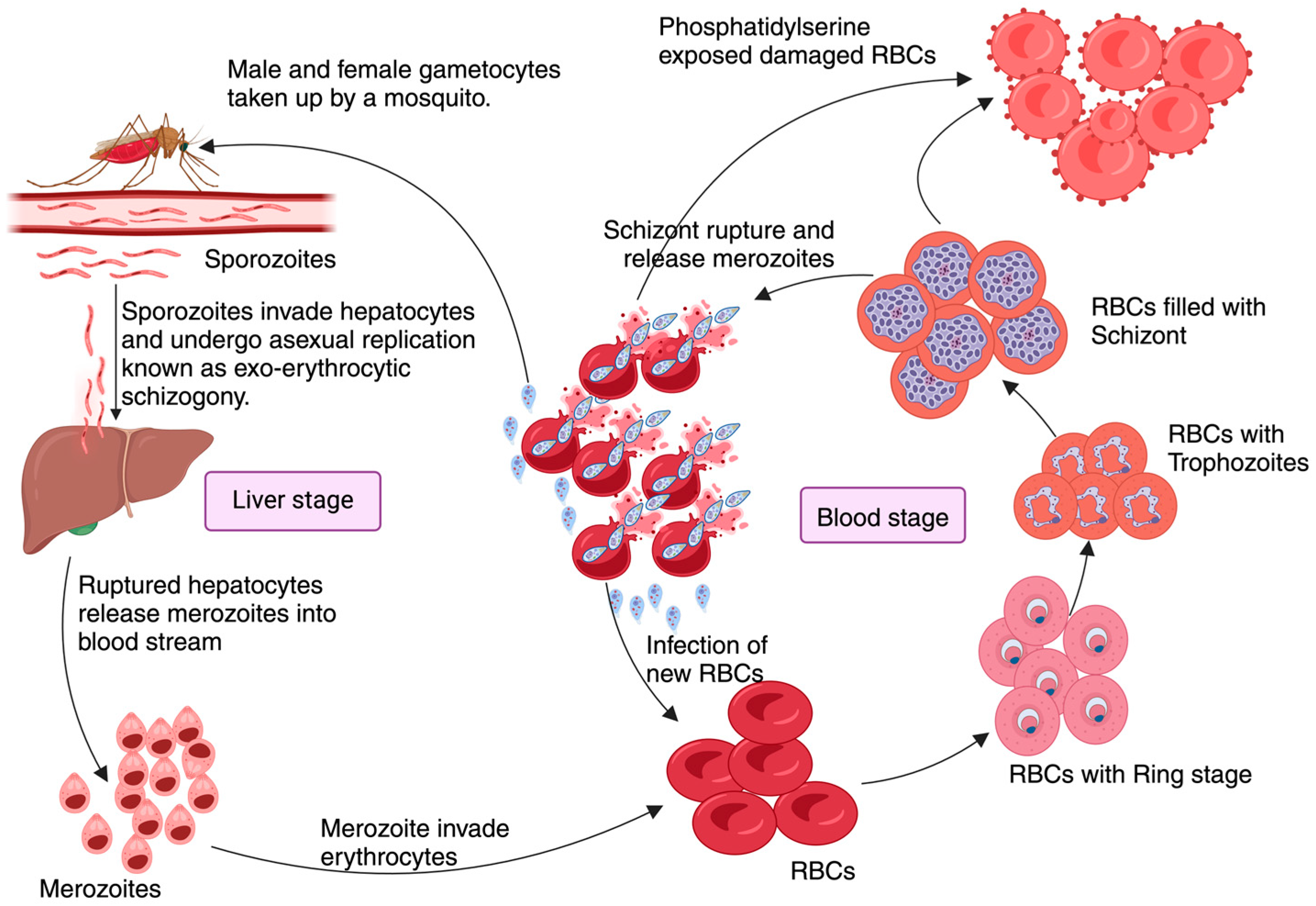
2. Pathophysiology of Malaria
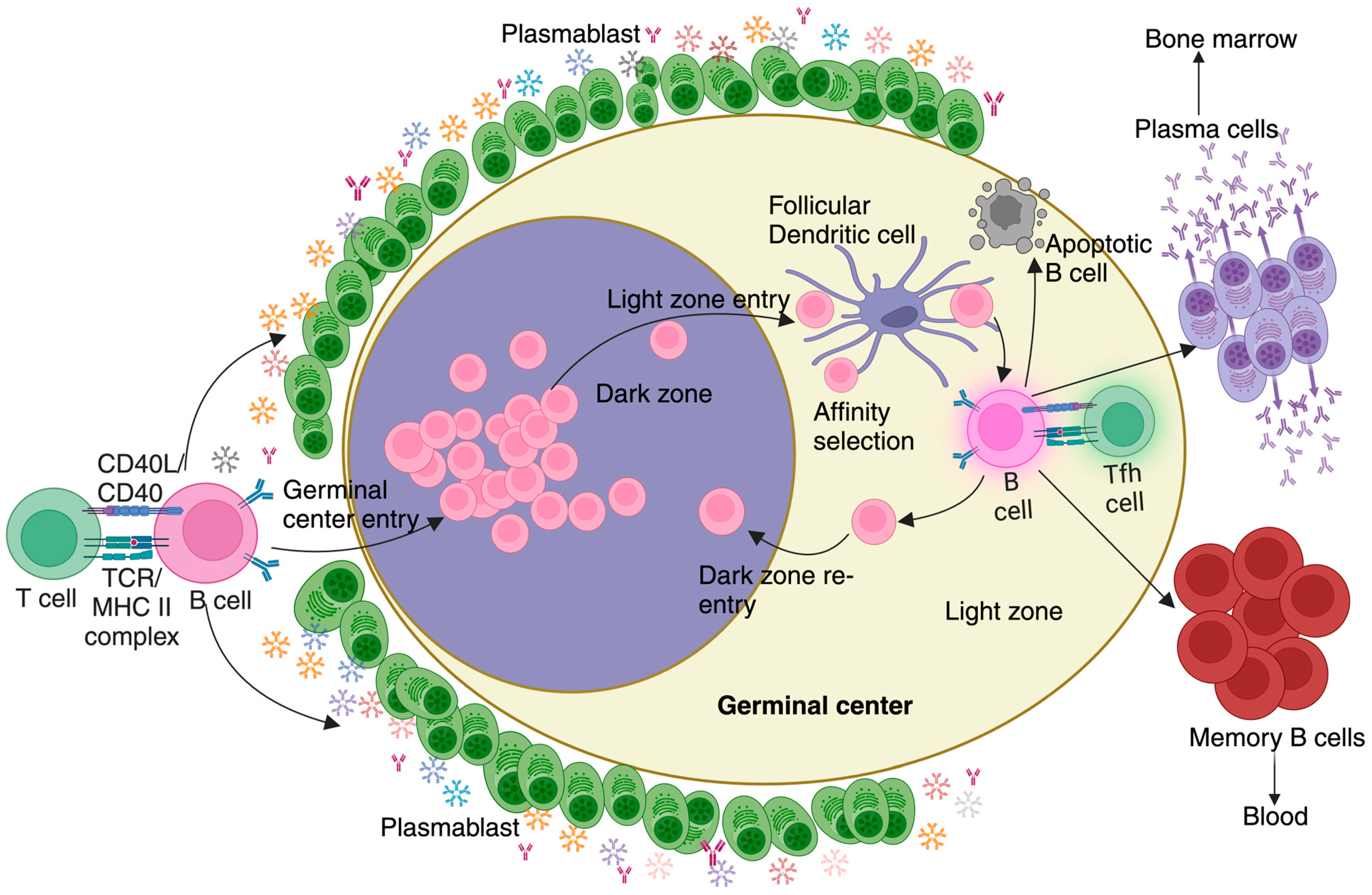
3. Anti-Malarial Immune Response
4. TAM Receptor Biology
5. TAM Receptors and Infections
5.1. AXL and Viral Infections
5.2. AXL and Malaria
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Milner, D.A., Jr. Malaria Pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Tran, T.M.; Li, S.; Doumbo, S.; Doumtabe, D.; Huang, C.Y.; Dia, S.; Bathily, A.; Sangala, J.; Kone, Y.; Traore, A.; et al. An intensive longitudinal cohort study of Malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clin. Infect. Dis. 2013, 57, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.L. Malaria’s Many Mates: Past, Present, and Future of the Systematics of the Order Haemosporida. J. Parasitol. 2014, 100, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, L.L.; Howard, R.J.; Aikawa, M.; Taraschi, T.F. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem. J. 1991, 274 Pt 1, 121–132. [Google Scholar] [CrossRef]
- Tokumasu, F.; Crivat, G.; Ackerman, H.; Hwang, J.; Wellems, T.E. Inward cholesterol gradient of the membrane system in P. falciparum-infected erythrocytes involves a dilution effect from parasite-produced lipids. Biol. Open 2014, 3, 529–541. [Google Scholar] [CrossRef]
- Hernandez-Castaneda, M.A.; Lavergne, M.; Casanova, P.; Nydegger, B.; Merten, C.; Subramanian, B.Y.; Matthey, P.; Lannes, N.; Mantel, P.Y.; Walch, M. A Profound Membrane Reorganization Defines Susceptibility of Plasmodium falciparum Infected Red Blood Cells to Lysis by Granulysin and Perforin. Front. Immunol. 2021, 12, 643746. [Google Scholar] [CrossRef]
- Vijay, R.; Guthmiller, J.J.; Sturtz, A.J.; Crooks, S.; Johnson, J.T.; Li, L.; Lan, L.Y.; Pope, R.L.; Chen, Y.; Rogers, K.J.; et al. Hemolysis-associated phosphatidylserine exposure promotes polyclonal plasmablast differentiation. J. Exp. Med. 2021, 218, e20202359. [Google Scholar] [CrossRef]
- Holz, L.E.; Fernandez-Ruiz, D.; Heath, W.R. Protective immunity to liver-stage malaria. Clin. Transl. Immunol. 2016, 5, e105. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Sack, B.K.; Baldwin, M.; Vaughan, A.M.; Kappe, S.H.I. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep. 2014, 7, 436–447. [Google Scholar] [CrossRef]
- Liehl, P.; Zuzarte-Luis, V.; Chan, J.; Zillinger, T.; Baptista, F.; Carapau, D.; Konert, M.; Hanson, K.K.; Carret, C.; Lassnig, C.; et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 2014, 20, 47–53. [Google Scholar] [CrossRef]
- Kurup, S.P.; Anthony, S.M.; Hancox, L.S.; Vijay, R.; Pewe, L.L.; Moioffer, S.J.; Sompallae, R.; Janse, C.J.; Khan, S.M.; Harty, J.T. Monocyte-Derived CD11c(+) Cells Acquire Plasmodium from Hepatocytes to Prime CD8 T Cell Immunity to Liver-Stage Malaria. Cell Host Microbe 2019, 25, 565–577 e566. [Google Scholar] [CrossRef] [PubMed]
- Angulo, I.; Fresno, M. Cytokines in the pathogenesis of and protection against malaria. Clin. Vaccine Immunol. 2002, 9, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Butcher, G.A.; Crandall, R.B. Action of malarial antibody in vitro. Nature 1969, 223, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Hodder, A.N.; Lin, C.S.; Hill, D.L.; Li Wai Suen, C.S.; Schofield, L.; Siba, P.M.; Mueller, I.; Cowman, A.F.; Hansen, D.S. Antibodies to the Plasmodium falciparum Proteins MSPDBL1 and MSPDBL2 Opsonize Merozoites, Inhibit Parasite Growth, and Predict Protection From Clinical Malaria. J. Infect. Dis. 2015, 212, 406–415. [Google Scholar] [CrossRef]
- Richards, J.S.; Stanisic, D.I.; Fowkes, F.J.; Tavul, L.; Dabod, E.; Thompson, J.K.; Kumar, S.; Chitnis, C.E.; Narum, D.L.; Michon, P.; et al. Association between naturally acquired antibodies to erythrocyte-binding antigens of Plasmodium falciparum and protection from malaria and high-density parasitemia. Clin. Infect. Dis. 2010, 51, e50–e60. [Google Scholar] [CrossRef]
- Cohen, S.; Mc, G.I.; Carrington, S. Gamma-globulin and acquired immunity to human malaria. Nature 1961, 192, 733–737. [Google Scholar] [CrossRef]
- Akter, J.; Khoury, D.S.; Aogo, R.; Lansink, L.I.M.; SheelaNair, A.; Thomas, B.S.; Laohamonthonkul, P.; Pernold, C.P.S.; Dixon, M.W.A.; Soon, M.S.F.; et al. Plasmodium-specific antibodies block in vivo parasite growth without clearing infected red blood cells. PLoS Pathog. 2019, 15, e1007599. [Google Scholar] [CrossRef]
- Kana, I.H.; Singh, S.K.; Garcia-Senosiain, A.; Dodoo, D.; Singh, S.; Adu, B.; Theisen, M. Breadth of Functional Antibodies Is Associated With Plasmodium falciparum Merozoite Phagocytosis and Protection Against Febrile Malaria. J. Infect. Dis. 2019, 220, 275–284. [Google Scholar] [CrossRef]
- Stephens, R.; Ndungu, F.M.; Langhorne, J. Germinal centre and marginal zone B cells expand quickly in a second Plasmodium chabaudi malaria infection producing mature plasma cells. Parasite Immunol. 2009, 31, 20–31. [Google Scholar] [CrossRef]
- Weiss, G.E.; Traore, B.; Kayentao, K.; Ongoiba, A.; Doumbo, S.; Doumtabe, D.; Kone, Y.; Dia, S.; Guindo, A.; Traore, A.; et al. The Plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010, 6, e1000912. [Google Scholar] [CrossRef] [PubMed]
- Wendel, B.S.; He, C.; Qu, M.; Wu, D.; Hernandez, S.M.; Ma, K.Y.; Liu, E.W.; Xiao, J.; Crompton, P.D.; Pierce, S.K.; et al. Accurate immune repertoire sequencing reveals malaria infection driven antibody lineage diversification in young children. Nat. Commun. 2017, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Vijay, R.; Guthmiller, J.J.; Sturtz, A.J.; Surette, F.A.; Rogers, K.J.; Sompallae, R.R.; Li, F.; Pope, R.L.; Chan, J.A.; de Labastida Rivera, F.; et al. Infection-induced plasmablasts are a nutrient sink that impairs humoral immunity to malaria. Nat. Immunol. 2020, 21, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Bockstal, V.; Guirnalda, P.; Caljon, G.; Goenka, R.; Telfer, J.C.; Frenkel, D.; Radwanska, M.; Magez, S.; Black, S.J. T. brucei infection reduces B lymphopoiesis in bone marrow and truncates compensatory splenic lymphopoiesis through transitional B-cell apoptosis. PLoS Pathog. 2011, 7, e1002089. [Google Scholar] [CrossRef] [PubMed]
- Obishakin, E.; de Trez, C.; Magez, S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. 2014, 36, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, M.; Guirnalda, P.; De Trez, C.; Ryffel, B.; Black, S.; Magez, S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008, 4, e1000078. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Ramonell, R.P.; Haddad, N.S.; Anam, F.A.; Rudolph, M.E.; Walker, T.A.; Truong, A.D.; Dixit, A.N.; Han, J.E.; Cabrera-Mora, M.; et al. Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature 2022, 611, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B.; Villoutreix, B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: Novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef]
- Hafizi, S.; Dahlback, B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006, 273, 5231–5244. [Google Scholar] [CrossRef]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 2016, 269, 44–59. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.K. Vitamin K-dependent gamma-glutamylcarboxylation: An ancient posttranslational modification. Vitam. Horm. 2008, 78, 157–184. [Google Scholar] [CrossRef] [PubMed]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Li, L.; Darden, T.; Monroe, D.M.; Pedersen, L.G. Prediction of solution structures of the Ca2+-bound gamma-carboxyglutamic acid domains of protein S and homolog growth arrest specific protein 6: Use of the particle mesh Ewald method. Biophys. J. 1997, 73, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Stafford, D.W. The vitamin K cycle. J. Thromb. Haemost. 2005, 3, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Mizuno, K. Roles of gamma-carboxylation and a sex hormone-binding globulin-like domain in receptor-binding and in biological activities of Gas6. FEBS Lett. 1997, 408, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P.; He, X.; Hardig, Y.; Dahlback, B.; Garcia de Frutos, P. Stimulation of Sky tyrosine phosphorylation by bovine protein S--domains involved in the receptor-ligand interaction. Eur. J. Biochem. 1997, 246, 147–154. [Google Scholar] [CrossRef]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hunter, T. Receptor signaling: When dimerization is not enough. Curr. Biol. 1999, 9, R568–R571. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Wiesmann, C.; Muller, Y.A.; de Vos, A.M. Ligand-binding sites in Ig-like domains of receptor tyrosine kinases. J. Mol. Med. 2000, 78, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Rajashankar, K.R.; Lackmann, M.; Cowan, C.A.; Henkemeyer, M.; Nikolov, D.B. Crystal structure of an Eph receptor-ephrin complex. Nature 2001, 414, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Heiring, C.; Dahlback, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Gohring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.A.; Kohrt, J.T.; Filipski, K.J.; Kaufman, M.; Sheehan, D.; Edmunds, J.E.; Delaney, A.; Wang, Y.; Bourbonais, F.; Lee, D.Y.; et al. Novel and selective spiroindoline-based inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2012, 22, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, C.; Simpson, C.; Deryckere, D.; Van Deusen, A.; Miley, M.J.; Kireev, D.; Norris-Drouin, J.; Sather, S.; Hunter, D.; et al. Discovery of Novel Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med. Chem. Lett. 2012, 3, 129–134. [Google Scholar] [CrossRef]
- Ling, L.; Templeton, D.; Kung, H.J. Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 18355–18362. [Google Scholar] [CrossRef]
- Brown, J.E.; Krodel, M.; Pazos, M.; Lai, C.; Prieto, A.L. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS ONE 2012, 7, e36800. [Google Scholar] [CrossRef]
- Adam-Artigues, A.; Arenas, E.J.; Arribas, J.; Prat, A.; Cejalvo, J.M. AXL—A new player in resistance to HER2 blockade. Cancer Treat. Rev. 2023, 121, 102639. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Krahling, S.; Callahan, M.K.; Williamson, P.; Schlegel, R.A. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999, 6, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Shiratsuchi, A.; Osada, S.; Kanazawa, S.; Nakanishi, Y. Essential role of phosphatidylserine externalization in apoptosing cell phagocytosis by macrophages. Biochem. Biophys. Res. Commun. 1998, 246, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, J.; Li, T.; Liu, C.; Bao, M.; Wang, X.; Li, X.; Li, J.; Huang, L.; Zhang, Z.; et al. Coreceptor AXL Facilitates African Swine Fever Virus Entry via Apoptotic Mimicry. J. Virol. 2023, 97, e0061623. [Google Scholar] [CrossRef]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.F.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.V.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef]
- Hastings, A.K.; Hastings, K.; Uraki, R.; Hwang, J.; Gaitsch, H.; Dhaliwal, K.; Williamson, E.; Fikrig, E. Loss of the TAM Receptor Axl Ameliorates Severe Zika Virus Pathogenesis and Reduces Apoptosis in Microglia. iScience 2019, 13, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL receptor is required for Zika virus strain MR-766 infection in human glioblastoma cell lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Zou, W.; Xu, P.; Cheng, F.; Zhang, E.Y.; Zhang-Chen, A.; Kleiboeker, S.; Qiu, J. Identification of AXL as a co-receptor for human parvovirus B19 infection of human erythroid progenitors. Sci. Adv. 2023, 9, eade0869. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Tay, E.S.; Shahidi, M.; O’Connor, K.S.; Booth, D.R.; George, J.; Douglas, M.W. Hepatitis C Virus Driven AXL Expression Suppresses the Hepatic Type I Interferon Response. PLoS ONE 2015, 10, e0136227. [Google Scholar] [CrossRef] [PubMed]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.Y.; Verma, S. Axl Promotes Zika Virus Entry and Modulates the Antiviral State of Human Sertoli Cells. mBio 2019, 10, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Zagorska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007, 196 (Suppl. S2), S259–S263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Y.; Wang, L.; You, X.; Li, J.; Zhang, G.; Hou, Y.; Wang, H.; He, S.; Li, E. Axl Mediates Resistance to Respiratory Syncytial Virus Infection Independent of Cell Attachment. Am. J. Respir. Cell Mol. Biol. 2022, 67, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Militerno, G.; Gugenheim, J.; Cuomo, O.; Hofman, P.; Mouiel, J.; Tovey, M. Synergistic interaction between anti-IFN alpha/beta antibody and low doses of cyclosporine therapy prolongs heart transplants in rats. Transplant. Proc. 1994, 26, 3050–3051. [Google Scholar]
- Shibata, T.; Makino, A.; Ogata, R.; Nakamura, S.; Ito, T.; Nagata, K.; Terauchi, Y.; Oishi, T.; Fujieda, M.; Takahashi, Y.; et al. Respiratory syncytial virus infection exacerbates pneumococcal pneumonia via Gas6/Axl-mediated macrophage polarization. J. Clin. Investig. 2020, 130, 3021–3037. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhen, Z.D.; Fan, D.Y.; Qin, C.F.; Han, D.S.; Zhou, H.N.; Wang, P.G.; An, J. Axl Deficiency Promotes the Neuroinvasion of Japanese Encephalitis Virus by Enhancing IL-1alpha Production from Pyroptotic Macrophages. J. Virol. 2020, 94, 10-1128. [Google Scholar] [CrossRef]
- Yang, J.; Li, M.; Yuan, M.; Bian, P.; Dong, Y.; Zhang, H.; Luo, C.; Xue, Z.; Wang, Y.; Zhang, F.; et al. Axl(−/−) neurons promote JEV infection by dampening the innate immunity. Virus Res. 2022, 307, 198605. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tian, L.; Yu, X.; Pattaradilokrat, S.; Li, J.; Wang, M.; Yu, W.; Qi, Y.; Zeituni, A.E.; Nair, S.C.; et al. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. USA 2014, 111, E511–E520. [Google Scholar] [CrossRef]
- Liu, D.; Winer, B.Y.; Chou, M.Y.; Tam, H.; Xu, Y.; An, J.; Gardner, J.M.; Cyster, J.G. Dynamic encounters with red blood cells trigger splenic marginal zone B cell retention and function. Nat. Immunol. 2024, 25, 142–154. [Google Scholar] [CrossRef]
- Urban, B.C.; Hien, T.T.; Day, N.P.; Phu, N.H.; Roberts, R.; Pongponratn, E.; Jones, M.; Mai, N.T.; Bethell, D.; Turner, G.D.; et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect. Immun. 2005, 73, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Achtman, A.H.; Khan, M.; MacLennan, I.C.; Langhorne, J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J. Immunol. 2003, 171, 317–324. [Google Scholar] [CrossRef]
- Weiss, L.; Geduldig, U.; Weidanz, W. Mechanisms of splenic control of murine malaria: Reticular cell activation and the development of a blood-spleen barrier. Am. J. Anat. 1986, 176, 251–285. [Google Scholar] [CrossRef]
- Hioki, A.; Yoshino, M.; Kano, S.; Ohtomo, H. Pathophysiology of hypoxia in mice infected with Plasmodium berghei. Parasitol. Res. 1987, 73, 298–302. [Google Scholar] [CrossRef]
- Park, M.K.; Ko, E.J.; Jeon, K.Y.; Kim, H.; Jo, J.O.; Baek, K.W.; Kang, Y.J.; Choi, Y.H.; Hong, Y.; Ock, M.S.; et al. Induction of Angiogenesis by Malarial Infection through Hypoxia Dependent Manner. Korean J. Parasitol. 2019, 57, 117–125. [Google Scholar] [CrossRef]
- Boeuf, P.; Tan, A.; Romagosa, C.; Radford, J.; Mwapasa, V.; Molyneux, M.E.; Meshnick, S.R.; Hunt, N.H.; Rogerson, S.J. Placental hypoxia during placental malaria. J. Infect. Dis. 2008, 197, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef] [PubMed]
- Blanco, Y.C.; Farias, A.S.; Goelnitz, U.; Lopes, S.C.; Arrais-Silva, W.W.; Carvalho, B.O.; Amino, R.; Wunderlich, G.; Santos, L.M.; Giorgio, S.; et al. Hyperbaric oxygen prevents early death caused by experimental cerebral malaria. PLoS ONE 2008, 3, e3126. [Google Scholar] [CrossRef] [PubMed]
- Hempel, C.; Combes, V.; Hunt, N.H.; Kurtzhals, J.A.; Grau, G.E. CNS hypoxia is more pronounced in murine cerebral than noncerebral malaria and is reversed by erythropoietin. Am. J. Pathol. 2011, 179, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Vandyke, K.; Zeissig, M.N.; Hewett, D.R.; Martin, S.K.; Mrozik, K.M.; Cheong, C.M.; Diamond, P.; To, L.B.; Gronthos, S.; Peet, D.J.; et al. HIF-2alpha Promotes Dissemination of Plasma Cells in Multiple Myeloma by Regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017, 77, 5452–5463. [Google Scholar] [CrossRef] [PubMed]
- Schoenhals, M.; Jourdan, M.; Bruyer, A.; Kassambara, A.; Klein, B.; Moreaux, J. Hypoxia favors the generation of human plasma cells. Cell Cycle 2017, 16, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Nalwoga, H.; Ahmed, L.; Arnes, J.B.; Wabinga, H.; Akslen, L.A. Strong Expression of Hypoxia-Inducible Factor-1alpha (HIF-1alpha) Is Associated with Axl Expression and Features of Aggressive Tumors in African Breast Cancer. PLoS ONE 2016, 11, e0146823. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.-Y.; Liu, Y.-W.; Zha, X.-N.; Tong, S.; Zhang, R.; He, X.-X.; Shan, S.-S.; Wang, K.; Liu, C.-Y. Overexpression of Axl reverses endothelial cells dysfunction in high glucose and hypoxia. J. Cell. Biochem. 2019, 120, 11831–11841. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
John, L.; Vijay, R. Role of TAM Receptors in Antimalarial Humoral Immune Response. Pathogens 2024, 13, 298. https://doi.org/10.3390/pathogens13040298
John L, Vijay R. Role of TAM Receptors in Antimalarial Humoral Immune Response. Pathogens. 2024; 13(4):298. https://doi.org/10.3390/pathogens13040298
Chicago/Turabian StyleJohn, Lijo, and Rahul Vijay. 2024. "Role of TAM Receptors in Antimalarial Humoral Immune Response" Pathogens 13, no. 4: 298. https://doi.org/10.3390/pathogens13040298
APA StyleJohn, L., & Vijay, R. (2024). Role of TAM Receptors in Antimalarial Humoral Immune Response. Pathogens, 13(4), 298. https://doi.org/10.3390/pathogens13040298