Effects of EGFR Inhibitor on Helicobacter pylori Induced Gastric Epithelial Pathology in Vivo


Abstract
:1. Introduction
2. Results
2.1. EKB-569 Effects on H. pylori-Induced Epithelial Responses in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EKB-569 nM | 0 | 1 nM | 10 nM | 100 nM | 1,000 nM |
|---|---|---|---|---|---|
| 700/800 nm RR | 65 ± 3 (100%) | 31 ± 10 * (48%) | 21 ± 7 ** (33%) | 19 ± 8 ** (29%) | 20 ± 5 ** (32%) |
2.2. H. pylori Infection of Mongolian Gerbils
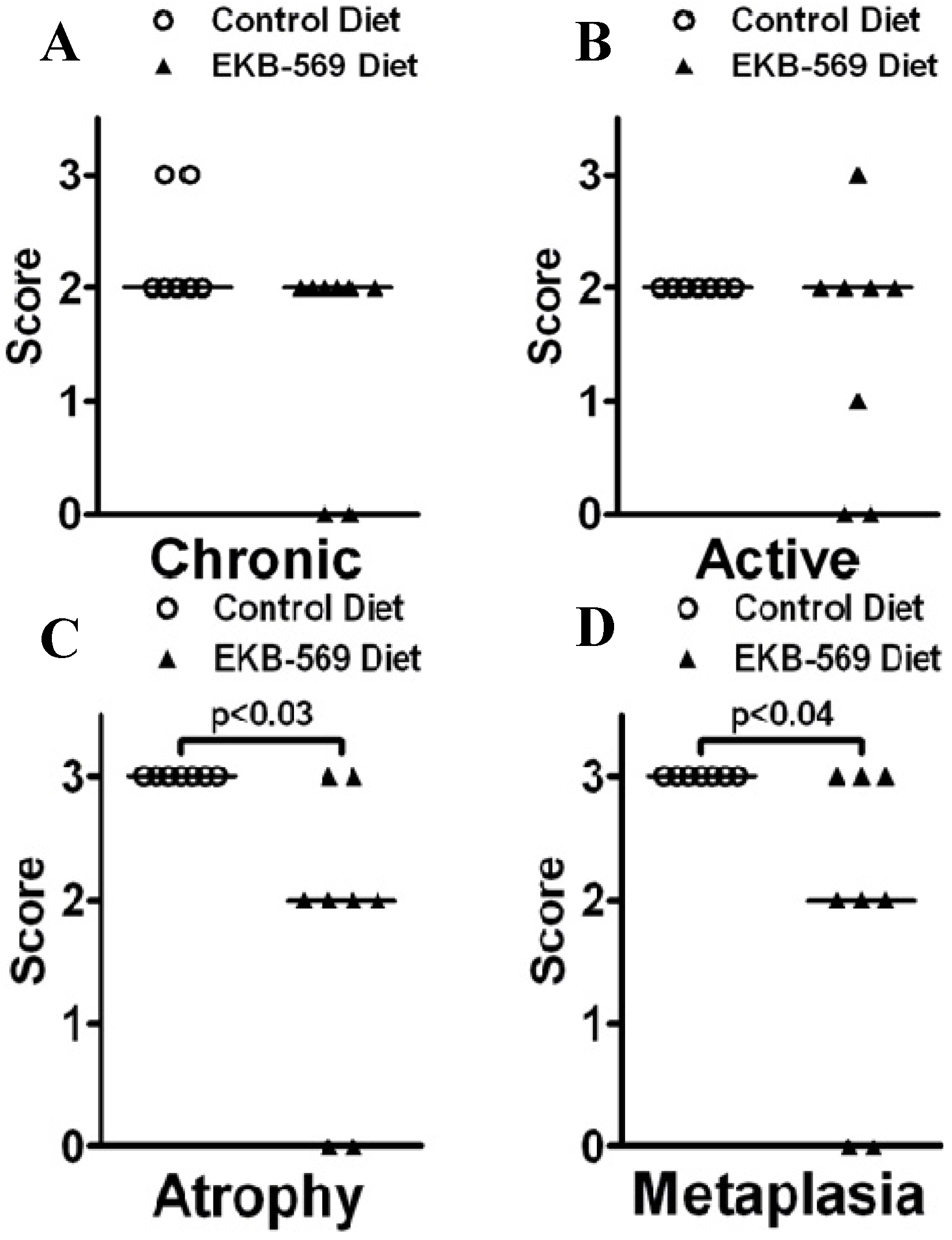
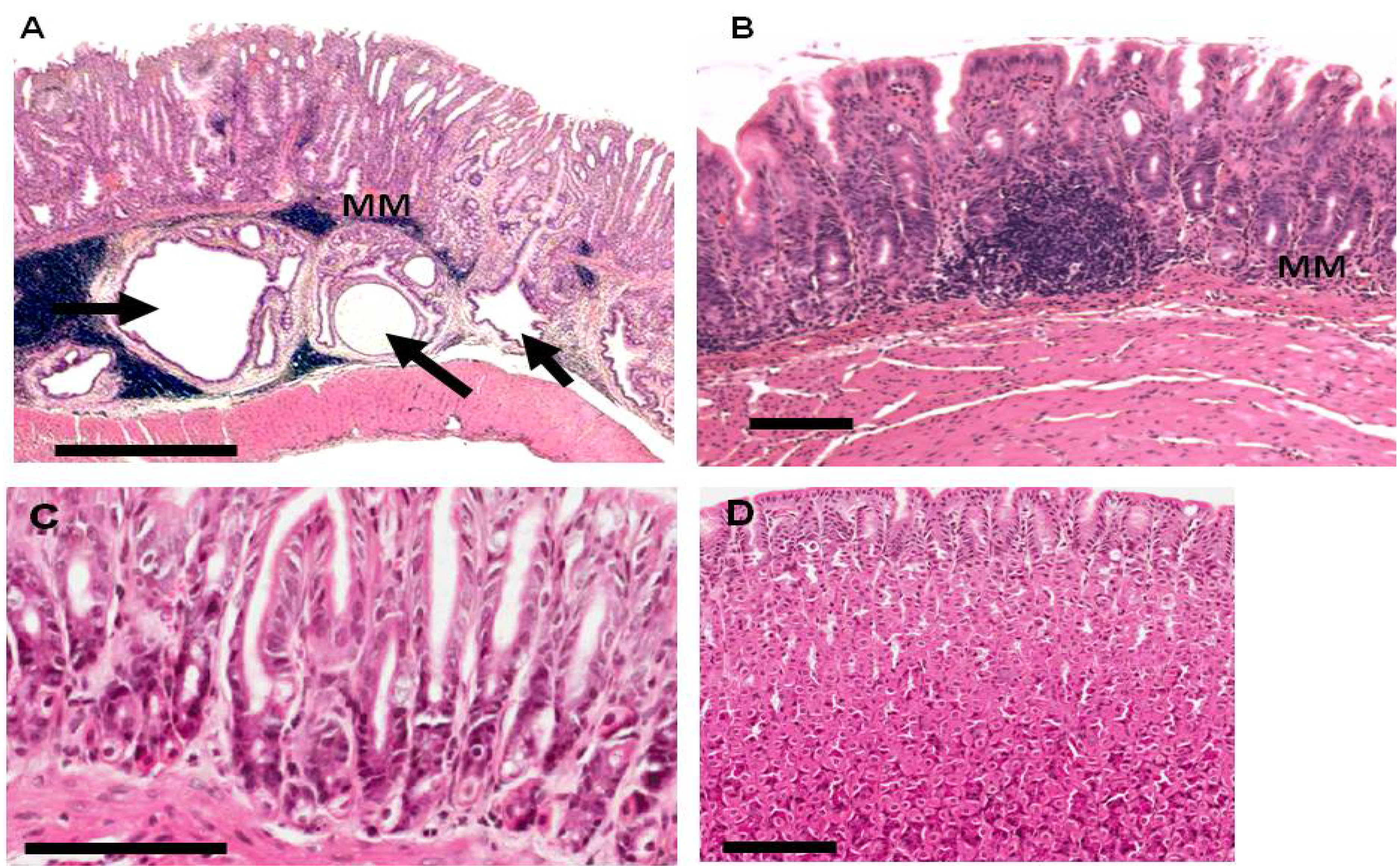
2.3. Gastric Pathology in EKB-569 Treated and Control H. pylori-Infected Gerbils


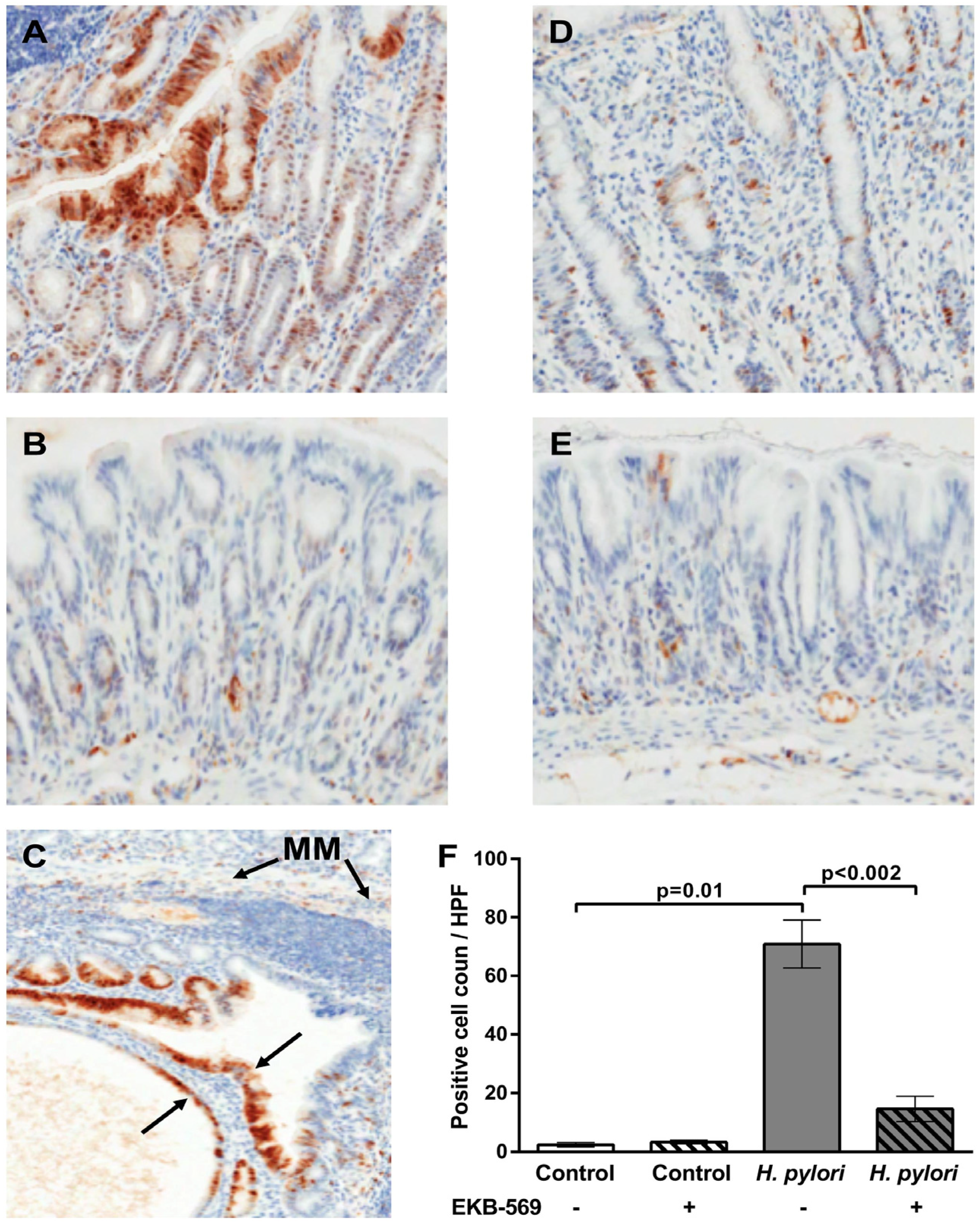
2.4. EGFR Inhibitor Reduces Phosphorylated Erk Positive Gastric Epithelial Cells in H. pylori-Infected Gerbils
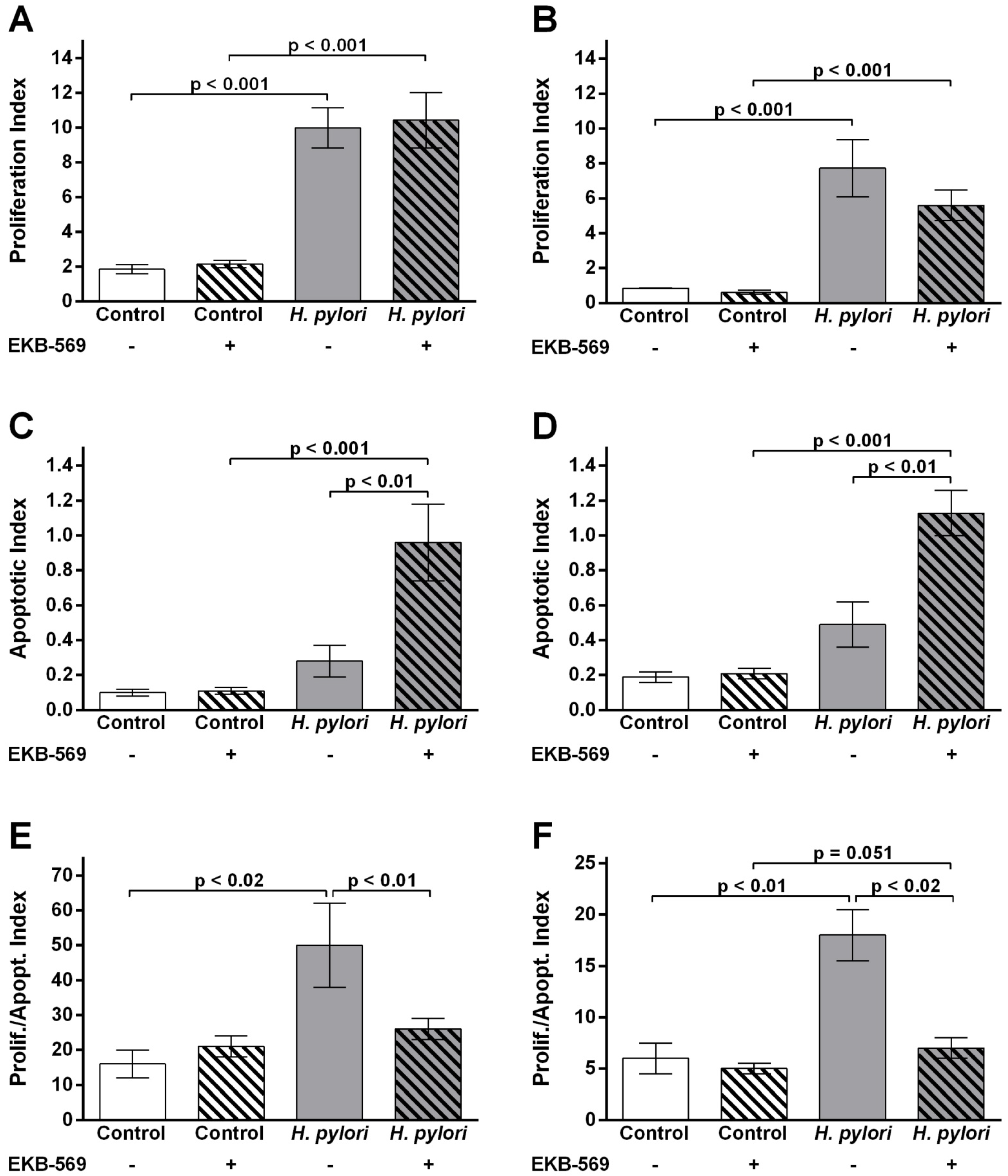
2.5. EGFR Inhibitor Modifies Gastric Epithelial Cell Proliferation Apoptosis Ratios in H. pylori- Infected Gerbils
2.6. Sequence Analysis of Gerbil Transcripts


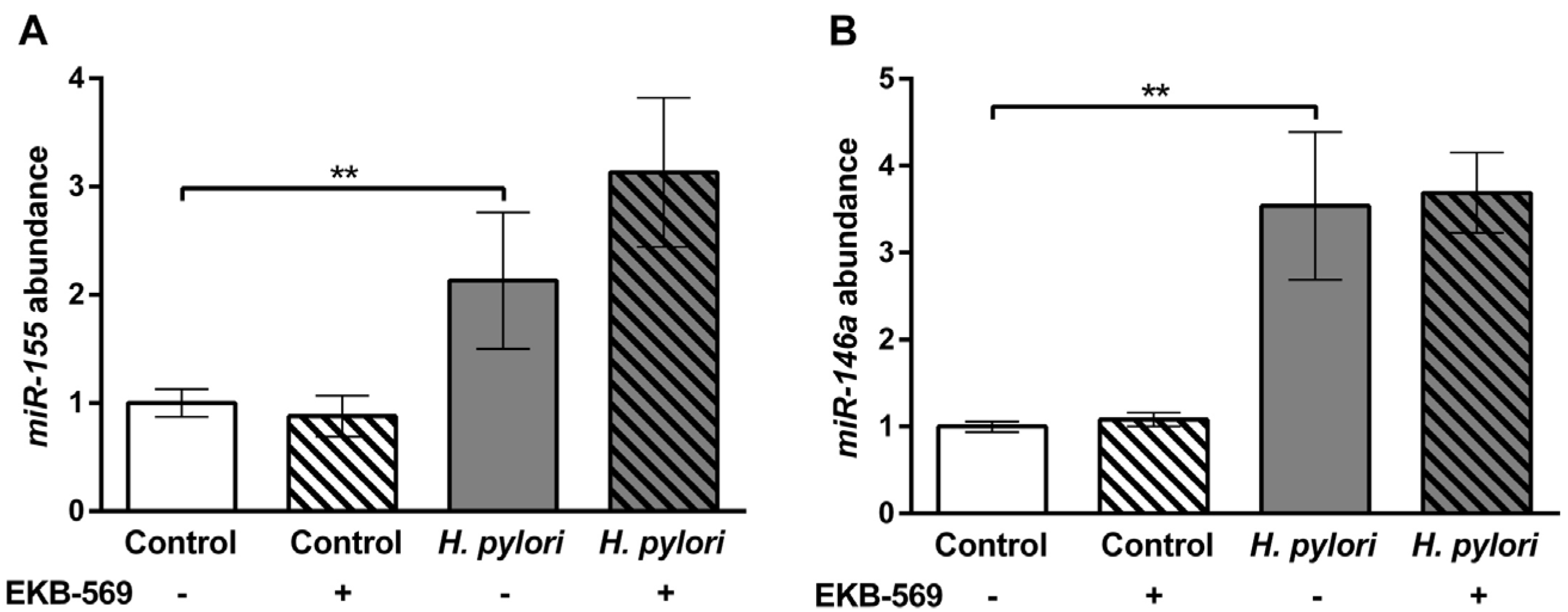
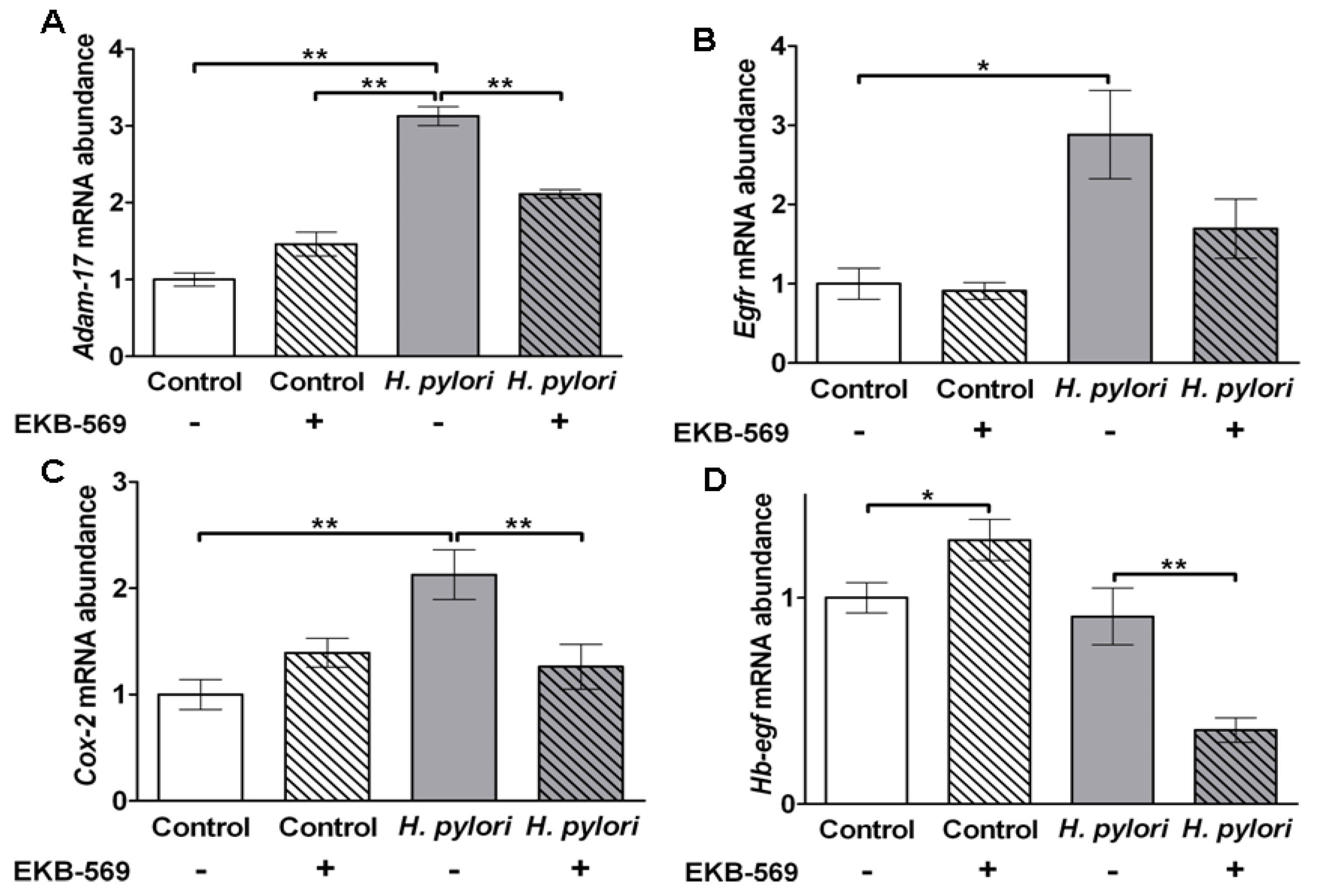
2.7.Effects of EGFR Inhibitor on H. pylori Induced Gastric Gene Expression in Gerbils


3. Discussion
4. Experimental
4.1. H. pylori Culture
4.2. In Vitro Bacterial-Epithelial Co-Culture
4.3. Infection of Mongolian Gerbils with H. pylori
4.4. Histological and Microbial Analysis of H. pylori Infection
4.5. Sequence Analysis of Mongolian Gerbil Genes
4.6. Quantitative Polymerase Chain Reaction (qPCR)
| Gene | Primers | Product size (bp) |
|---|---|---|
| Adam17 | 5'-AAAGGGAACCCTGTACCGTAGGG 5'-GCCAAAAACTTTCCGAAAGTGT | 131 |
| Cox-2 | 5'-AGTCTCTCAACGAATACCGCAAAC5'-ATGTCACTGTAGAGGGCTTTCAAC | 117 |
| Egfr | 5'-GGGAAATGCTCTGTACGAAAACAC5'-AGCACCGGTCAGGATTTCCT | 118 |
| Gapdh | 5'-CCTGTGACTTTAACAGCGACTCC5'-CCATGAGGTCCACCACCCT | 102 |
| Hb-egf | 5'-TCGGAGAGGTCTGGCGG5'-TCCTGGACTTCCTGAGTGCG | 118 |
| Ifn-γ | 5'-CCATGAACGCTACACACTGCATC5'-GAAGTAGAAAGAGACAATCTGG | 230 |
| miRNAs | Forward Primers | |
| U6 snRNA | 5'-dTGGCCCCTGCGCAAGGATG | - |
| miR-146a | 5'-dTGAGAACTGAATTCCATGGGTT | - |
| miR-155 | 5'-dTTAATGCTAATTGTGATAGGGGT | - |
4.7. Quantitative PCR Analysis of miRNAs Expression in Gerbil Gastric Mucosa
4.8. Statistics
5. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Peek, R.M.; Crabtree, J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248. [Google Scholar] [CrossRef]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2012, 10, 403–414. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Naumann, M. Epithelial cell signalling in Helicobacter pylori infection. Curr. Signal Transduct. Ther. 2006, 1, 53–65. [Google Scholar] [CrossRef]
- Backert, S.; Naumann, M. What a disorder: Proinflammatory signalling pathways induced by Helicobacter pylori. Trends Microbiol. 2010, 18, 479–486. [Google Scholar] [CrossRef]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef]
- Peek, R.M.; Moss, S.F.; Tham, K.T.; Pérez-Pérez, G.I.; Wang, S.; Miller, G.G.; Atherton, J.C.; Holt, P.R.; Blaser, M.J. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation and apoptosis. J. Natl. Cancer Inst. 1997, 89, 863–868. [Google Scholar] [CrossRef]
- Moss, S.F.; Sordillo, E.M.; Abdalla, A.M.; Makarov, V.; Hanzely, Z.; Perez-Perez, G.I.; Blaser, M.J.; Holt, P.R. Increased gastric epithelial cell apoptosis associated with colonisation with cagA+ H. pylori strains. Cancer Res. 2001, 61, 1406–1411. [Google Scholar]
- Peek, R.M.; Wirth, H.P.; Moss, S.F.; Yang, M.; Abdalla, A.M.; Tham, K.T.; Zhang, T.; Tang, L.H.; Modlin, I.M.; Blaser, M.J. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology 2000, 118, 48–59. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Court, M.; Aboshkiwa, M.A.; Jeremy, A.H.T.; Dixon, M.F.; Robinson, P.A. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J. Pathol. 2004, 202, 197–207. [Google Scholar] [CrossRef]
- Court, M.; Robinson, P.A.; Dixon, M.F.; Jeremy, A.H.T.; Crabtree, J.E. The effect of gender on Helicobacter felis mediated gastritis, epithelial cell proliferation and apoptosis in the mouse model. J. Pathol. 2003, 201, 303–311. [Google Scholar] [CrossRef]
- Court, M.; Robinson, P.A.; Dixon, M.F.; Crabtree, J.E. Gastric Helicobacter infection in murine and gerbil models: comparative analysis of effects of H. pylori and H. felis on gastric epithelial cell proliferation. J. Infect. Dis. 2002, 186, 1348–1352. [Google Scholar] [CrossRef]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef]
- Wallasch, C.; Crabtree, J.E.; Bevac, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 295, 695–701. [Google Scholar] [CrossRef]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M.; Shaw, L.M.; Kelly, C.P. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 2001, 276, 48127–48134. [Google Scholar]
- Du, Y.; Danjo, K.; Robinson, P.A.; Crabtree, J.E. In-Cell Western analysis of Helicobacter pylori induced phosphorylation of extracelluar-signal related kinase via the transactivation of the epidermal growth factor receptor. Microbes Infect. 2007, 9, 838–846. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wang, J.; Dong, Z.; Mian, S.; Yu, F.X. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 2004, 45, 2569–2576. [Google Scholar] [CrossRef]
- Yan, F.; Cao, H.; Chaturvedi, R.; Krishna, U.; Hobbs, S.S.; Dempsey, P.J.; Peek, R.M., Jr.; Cover, T.L.; Washington, M.K.; Wilson, K.T.; et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 2009, 136, 1297–1307. [Google Scholar] [CrossRef]
- Yoshimura, T.; Tomita, T.; Dixon, M.F.; Axon, A.T.R.; Robinson, P.A.; Crabtree, J.E. ADAMs mRNA expression in Helicobacter pylori infected, normal and neoplastic gastric mucosa. J. Infect. Dis. 2002, 185, 332–340. [Google Scholar] [CrossRef]
- Cox, J.M.; Clayton, C.L.; Tomita, T.; Wallace, D.M.; Robinson, P.A.; Crabtree, J.E. cDNA array analysis of cag pathogenicity island-associated H. pylori epithelial cell response genes. Infect. Immun. 2001, 69, 6970–6980. [Google Scholar] [CrossRef]
- Wong, B.C.; Wang, W.P.; So, W.H.; Shin, V.Y.; Wong, W.M.; Fung, F.M.; Liu, E.S.; Hiu, W.M.; Lam, S.K.; Cho, C.H. Epidermal growth factor and its receptor in chronic active gastritis and gastroduodenal ulcer before and after H. pylori eradication. Aliment. Pharmacol. Ther. 2001, 15, 1459–1465. [Google Scholar] [CrossRef]
- Naef, M.; Yokoyama, M.; Friess, H.; Buchler, M.W.; Korc, M. Co-expression of heparin-binding EGF-like growth factor and related peptides in human gastric carcinoma. Int. J. Cancer 1996, 66, 315–321. [Google Scholar] [CrossRef]
- Keates, S.; Keates, A.C.; Katchar, K.; Peek, R.M., Jr.; Kelly, C.P. Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Infect. Dis. 2007, 196, 95–103. [Google Scholar] [CrossRef]
- Bauer, B.; Bartfeld, S.; Meyer, T. H. pylori selectively blocks EGFR endocytosis via the non-receptor kinase c-Abl and CagA. Cell. Microbiol. 2009, 11, 156–169. [Google Scholar] [CrossRef]
- Tabassam, F.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori activate epidermal growth factor receptor-and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3β. Cell. Microbiol. 2009, 11, 70–82. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Zabler, D.; Schmidt, D.; Hartig, R.; Brandt, S.; Backert, S. Importance of EGFR receptor: HER2/Neu and Erk 1/2 kinase signalling for host cell elongation and scattering induced by Helicobacter pylori CagA protein: Antagonistic effects of the vacuoalting cytotoxin VacA. Cell. Microbiol. 2009, 11, 488–505. [Google Scholar] [CrossRef]
- Wang, J.; Court, M.; Jeremy, A.H.T.; Aboshkiwa, M.A.; Robinson, P.A.; Crabtree, J.E. Infection of Mongolian gerbils with Chinese Helicobacter pylori strains. FEMS Immunol. Med. Microbiol. 2003, 36, 207–213. [Google Scholar] [CrossRef]
- Watananabe, T.; Tada, M.; Nagai, H.; Sasaki, S.; Nakao, M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998, 115, 642–648. [Google Scholar] [CrossRef]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef]
- Torrance, C.J.; Jackson, P.E.; Montgomery, E.; Kinzler, K.W.; Vogelstein, B.; Wissner, A.; Nunes, M.; Frost, P.; Discafani, C.M. Combinatorial chemoprevention of intestinal neoplasia. Nat. Med. 2000, 6, 1024–1028. [Google Scholar] [CrossRef]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Xiao, B.; Liu, Z.; Li, B.S.; Tang, B.; Li, W.; Guo, G.; Shi, Y.; Wang, F.; Wu, Y.; Tong, W.D.; et al. Induction of microRNA-155 during Helicobacter pylori infection and it’s negative regulatory role in the inflammatory response. J. Infect. Dis. 2009, 200, 916–925. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, B.; Tang, B.; Li, B.; Li, N.; Zhu, E.; Guo, G.; Gu, J.; Zhuang, Y.; Liu, X.; et al. Up-regulated microRNA-146a negatively modulate Helicobacter pylori-induced inflammatory response in human gastric epithelial cells. Microbes Infect. 2010, 12, 854–863. [Google Scholar] [CrossRef]
- Busiello, I.; Acquaviva, R.; di Popolo, A.; Blanchard, T.G.; Ricci, V.; Romano, M.; Zarrilli, R. Helicobacter pylori γ-glutamyltranspeptidase upregulates Cox-2 and EGF-related peptide expression in human gastric cells. Cell. Microbiol. 2004, 6, 255–267. [Google Scholar] [CrossRef]
- Beales, I.L.P. Gastrin and interleukin-1 beta stimulate growth factor secretion from cultured rabbit gastric parietal cells. Life Sci. 2004, 75, 2983–2995. [Google Scholar] [CrossRef]
- Varro, A.; Noble, P.J.; Wroblewski, L.E.; Bishop, L.; Dockray, G.J. Gastrin-cholecystokininB receptor expression in AGS cells is associated with direct inhibition and indirect stimulation of cell proliferation via paracrine activation of the epidermal growth factor receptor. Gut 2002, 50, 827–833. [Google Scholar] [CrossRef]
- Tanida, S.; Joh, T.; Itoh, K.; Kataoka, H.; Sasaki, M.; Ohara, H.; Nakazawa, T.; Nomura, T.; Kinugasa, Y.; Ohmoto, H.; et al. The mechanisms of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology 2004, 127, 559–569. [Google Scholar] [CrossRef]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 tranactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 2002, 8, 289–293. [Google Scholar] [CrossRef]
- Wissner, A.; Overbeek, E.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Mamuya, N.; Rosfjord, E.C.; Discafani, C.; Davis, R.; Shi, X.; et al. Synthesis and structure activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the EGFR and human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 2002, 46, 49–63. [Google Scholar]
- Naumann, M.; Crabtree, J.E. Helicobacter pylori-induced epithelial signalling in gastric carcinogenesis. Trends Microbiol. 2004, 12, 29–36. [Google Scholar] [CrossRef]
- Khokhlatchev, A.V.; Canagarajah, B.; Wilsbacher, J.; Robinson, M.; Atkinson, M.; Goldsmith, E.; Cobb, M.H. Phosphorylation of the MAP Kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 1998, 93, 605–615. [Google Scholar] [CrossRef]
- Playford, R.J.; Hanby, A.M.; Gschmeissner, S.; Peiffer, L.P.; Wright, N.A.; McGarrity, T. The epidermal growth factor receptor (EGF-R) is present on the basolateral, but not the apical, surface of enterocytes in the human gastrointestinal tract. Gut 1996, 39, 262–266. [Google Scholar] [CrossRef]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. USA 2009, 106, 22369–22374. [Google Scholar] [CrossRef]
- Kajanne, R.; Miettinen, P.; Mehlem, A.; Leivonen, S.K.; Birrer, M.; Foschi, M.; Kahari, V.M.; Leppa, S. EGF-R regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell. Physiol. 2007, 212, 489–497. [Google Scholar] [CrossRef]
- McCaig, C.; Duval, C.; Hemers, E.; Steele, I.; Pritchard, D.M.; Przemeck, S.; Dimaline, R.; Ahmed, S.; Bodger, K.; Kerrigan, D.D.; et al. The role of matrix metalloproteinase-7 in redefining the gastric microenvironment in response to Helicobacter pylori. Gastroenterology 2006, 130, 1754–1763. [Google Scholar] [CrossRef]
- Tyner, J.W.; Kim, E.Y.; Ide, K.; Pelletier, M.R.; Roswitt, W.T.; Morton, J.D.; Battaile, J.T.; Patel, A.C.; Patterson, G.A.; Castro, M.; et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J. Clin. Invest. 2006, 116, 309–321. [Google Scholar] [CrossRef]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009, 5, e1000603. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Ferrero, R.; Kusters, H. The mouse colonising Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 2002, 7, 139–140. [Google Scholar] [CrossRef]
- Fu, S.; Ramanujam, K.S.; Wong, A.; Fantry, G.T.; Drachenberg, C.B.; James, S.P.; Meltzer, S.J.; Wilson, K.T. Increased expression and cellular localization of inducible nitric oxide synthase and cyloxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 1999, 116, 1319–1329. [Google Scholar] [CrossRef]
- Caputo, R.; Tuccillo, C.; Manzo, B.A.; Zarrilli, R.; Tortora, G.; Blanco, C.V.; Ricci, V.; Ciardiello, F.; Romano, M. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric epithelial cells through an epidermal growth factor receptor-, cycclooxygenase-2 dependent mechanism. Clin. Cancer Res. 2003, 9, 2015–2021. [Google Scholar]
- Sierra, J.C.; Hobbs, S.; Chaturvedi, R.; Yan, F.; Wilson, K.T.; Peek, R.M., Jr.; Polk, B.R. Induction of COX-2 expression by Helicobacter pylori is mediated by activation of epidermal growth factor receptor in gastric epithelial cells. Am. J. Physiol. 2013, 305, G196–G203. [Google Scholar]
- Wu, C.Y.; Wu, M.S.; Kuo, K.N.; Wang, C.B.; Chen, Y.J.; Lin, J.T. Effective reduction of gastric cancer risk with regular use of nonsteroidal anti-inflammatory drugs in Helicobacter pylori-infected patients. J. Clin. Oncol. 2010, 28, 2952–2957. [Google Scholar] [CrossRef]
- Tomtitchong, P. Host Epithelial Cell Signaling in Gastric Helicobacter pylori Infection. Ph.D. Thesis, University of Leeds, Leeds, UK, 2008. [Google Scholar]
- Zanella, C.L.; Posada, P.; Tritton, T.R.; Mossman, B.T. Asbestos causes stimulation of the extracellular signal-regulated kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res. 1996, 56, 5334–5338. [Google Scholar]
- Manning, C.B.; Cummins, A.B.; Jung, M.W.; Berlanger, I.; Timblin, C.R.; Palmer, C.; Taatjes, D.J.; Hemenway, D.; Vacek, P.; Mossman, B.T. A mutant epidermal growth factor receptor targeted to lung epithelium inhibits asbestos-induced proliferation and proto-oncogene expression. Cancer Res. 2002, 62, 4169–4175. [Google Scholar]
- Roth, R.A.; Kapadia, S.B.; Martin, S.M.; Lorenz, R.G. Cellular immune responses are essential for development of Helicobacter felis associated gastric pathology. J. Immunol. 1999, 163, 1490–1497. [Google Scholar]
- Shibita, W.; Hirata, Y.; Maeda, S.; Oguru, K.; Yania, A.; Mitsuno, Y.; Yamaji, Y.; Okamoto, M.; Yoshida, H.; Kawabe, T.; et al. CagA protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the Mongolian gerbil model. J. Pathol. 2006, 210, 306–314. [Google Scholar] [CrossRef]
- Grimminger, F.; Schermuly, R.T.; Ghofrani, H.A. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat. Rev. Drug Discov. 2010, 9, 956–970. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Crabtree, J.E.; Jeremy, A.H.T.; Duval, C.; Dixon, M.F.; Danjo, K.; Carr, I.M.; Pritchard, D.M.; Robinson, P.A. Effects of EGFR Inhibitor on Helicobacter pylori Induced Gastric Epithelial Pathology in Vivo. Pathogens 2013, 2, 571-590. https://doi.org/10.3390/pathogens2040571
Crabtree JE, Jeremy AHT, Duval C, Dixon MF, Danjo K, Carr IM, Pritchard DM, Robinson PA. Effects of EGFR Inhibitor on Helicobacter pylori Induced Gastric Epithelial Pathology in Vivo. Pathogens. 2013; 2(4):571-590. https://doi.org/10.3390/pathogens2040571
Chicago/Turabian StyleCrabtree, Jean E., Anthony H.T. Jeremy, Cedric Duval, Michael F. Dixon, Kazuma Danjo, Ian M. Carr, D. Mark Pritchard, and Philip A. Robinson. 2013. "Effects of EGFR Inhibitor on Helicobacter pylori Induced Gastric Epithelial Pathology in Vivo" Pathogens 2, no. 4: 571-590. https://doi.org/10.3390/pathogens2040571
APA StyleCrabtree, J. E., Jeremy, A. H. T., Duval, C., Dixon, M. F., Danjo, K., Carr, I. M., Pritchard, D. M., & Robinson, P. A. (2013). Effects of EGFR Inhibitor on Helicobacter pylori Induced Gastric Epithelial Pathology in Vivo. Pathogens, 2(4), 571-590. https://doi.org/10.3390/pathogens2040571