Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection


Abstract
:
1. Introduction
2. Results
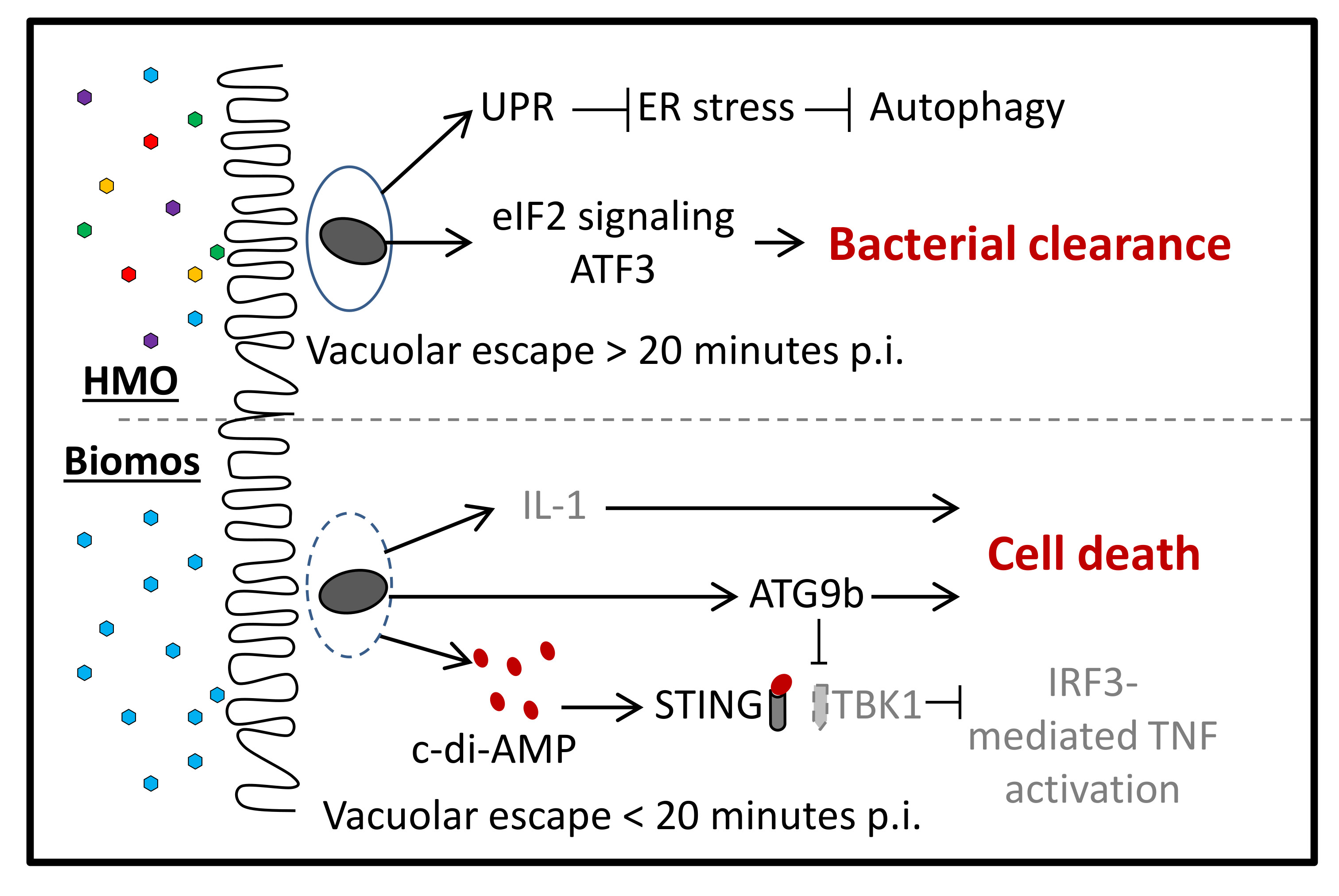
2.1. HMO Pretreatment Reduces L. monocytogenes Host Invasion
2.2. Prebiotics Modify L. monocytogenes Infection Dynamics
2.3. Biomos Drives Differential Expression of L. monocytogenes Genes during Infection
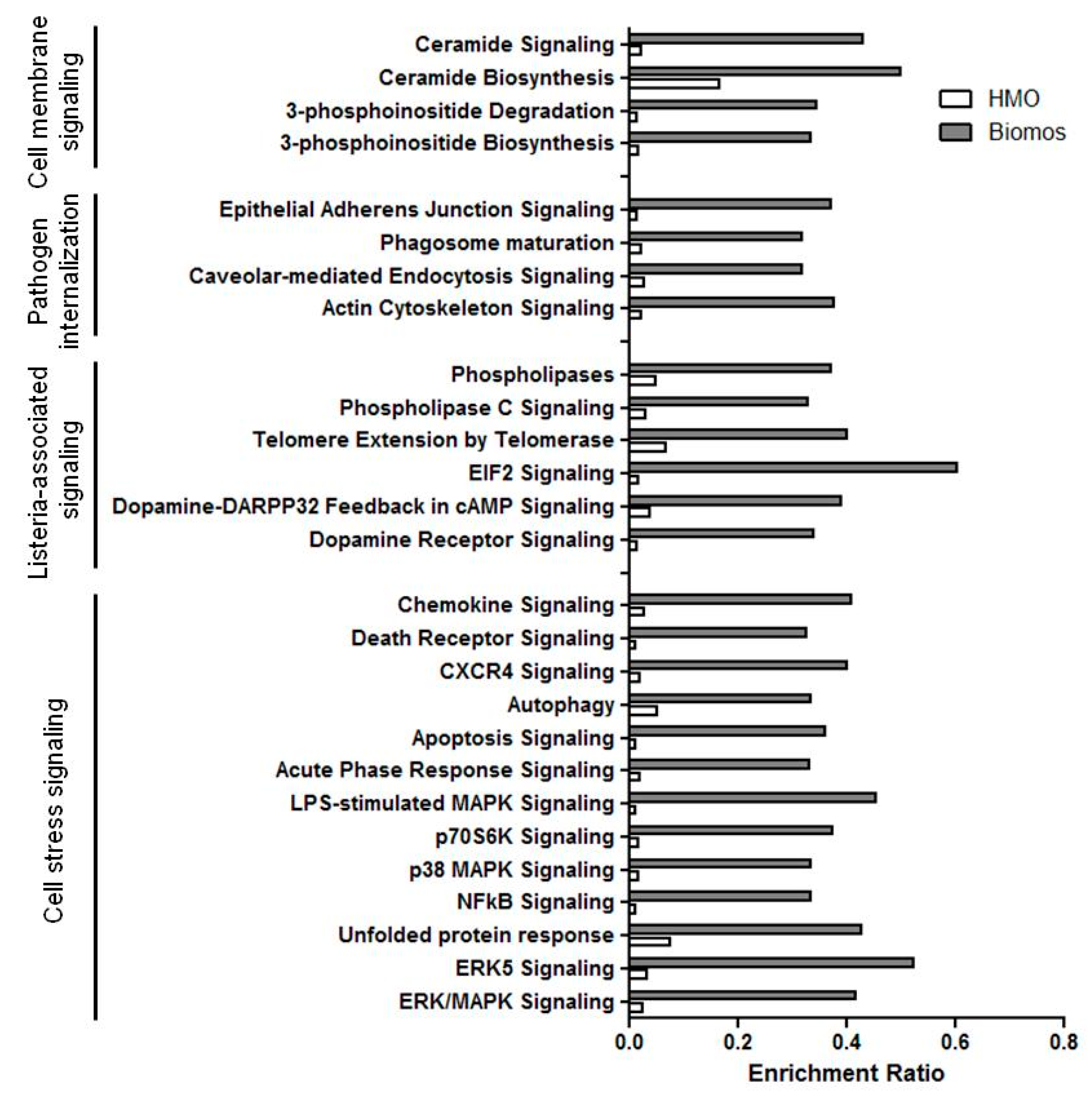
3. Differential Gene Set Enrichment of Host Cell Signaling Pathways
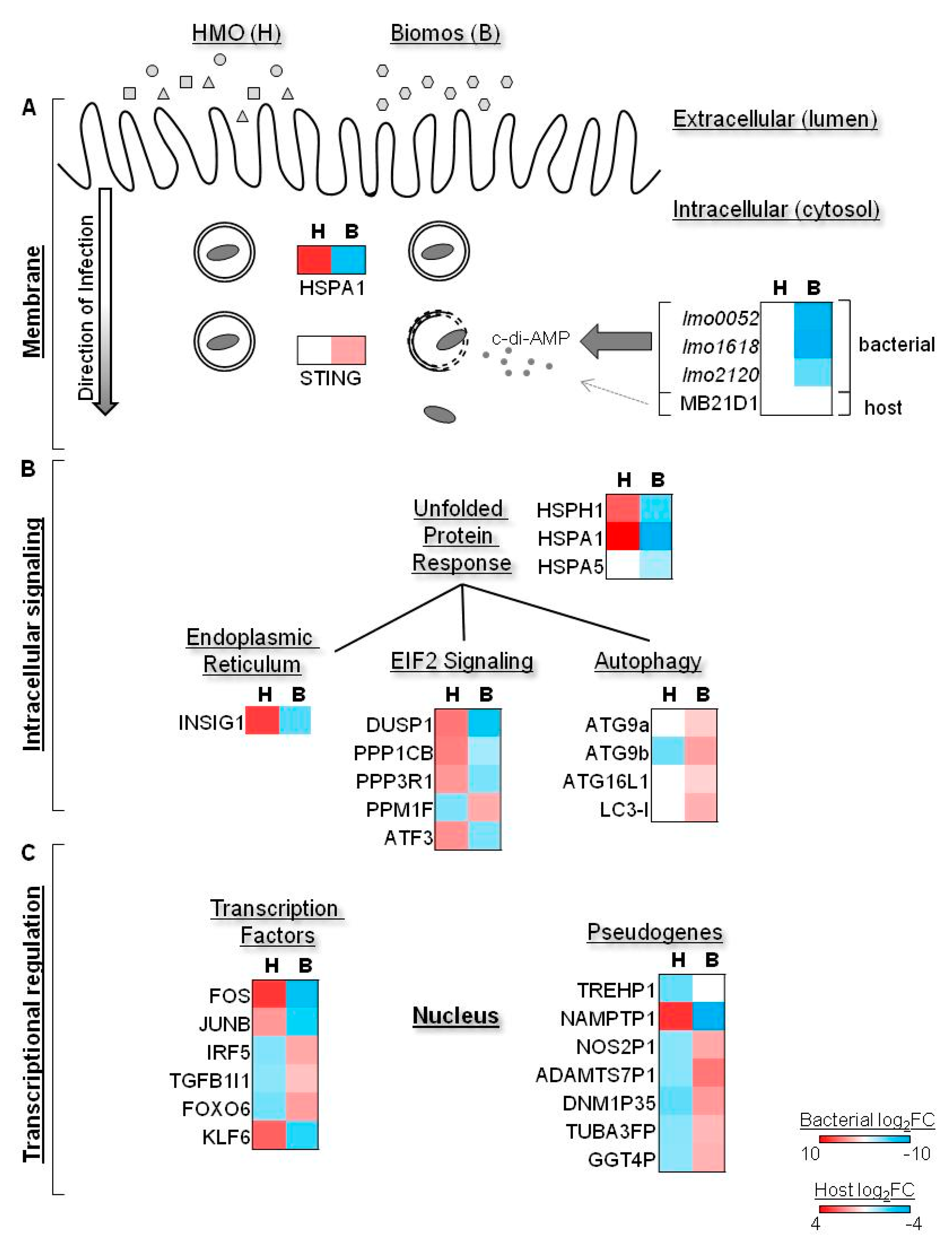
4. Prebiotic-Dependent Differential Expression of Host Cell Stress Signaling during Infection
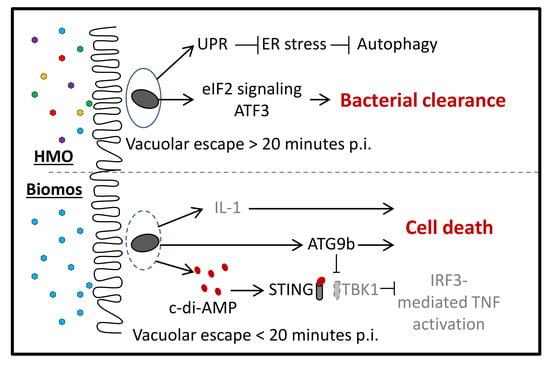
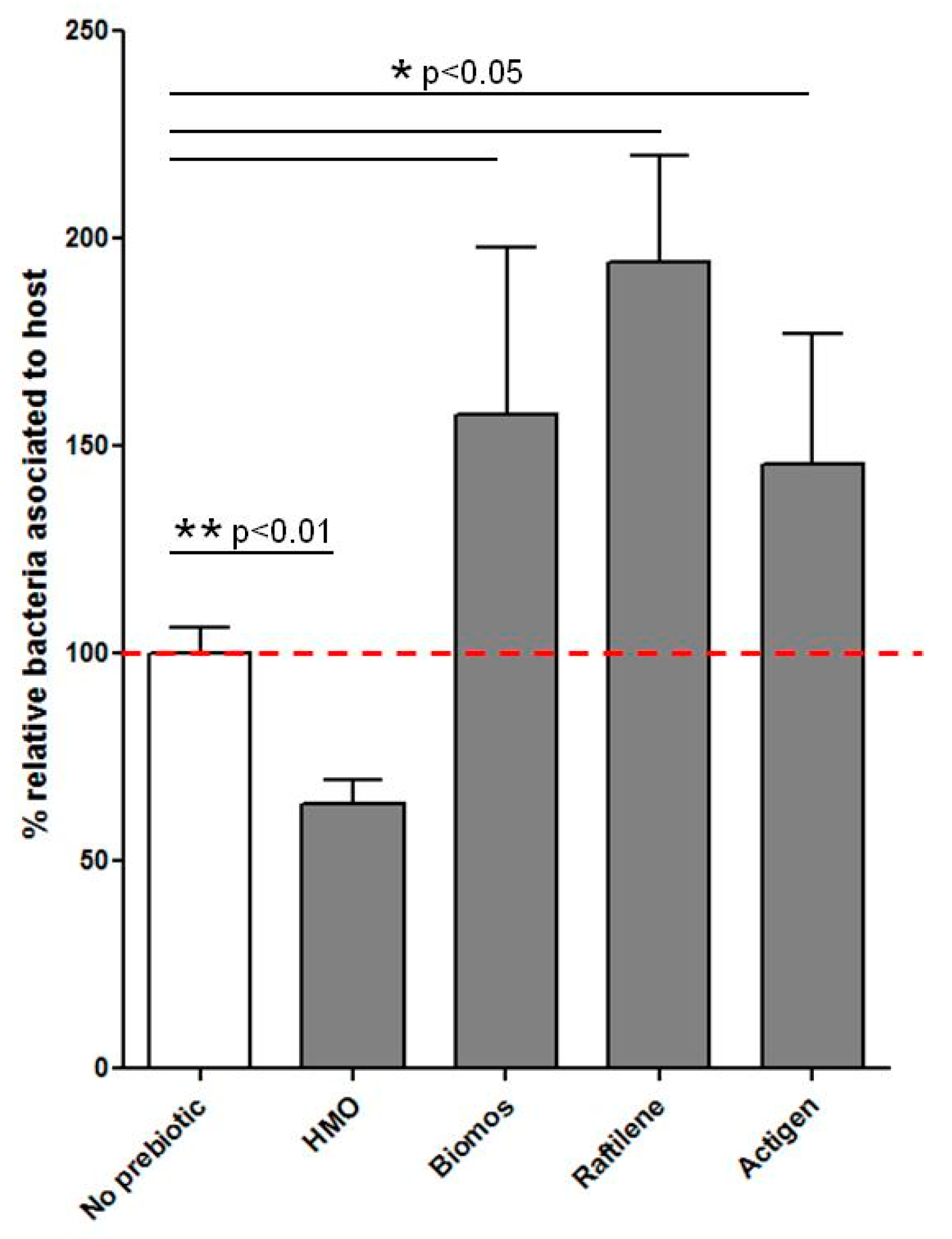
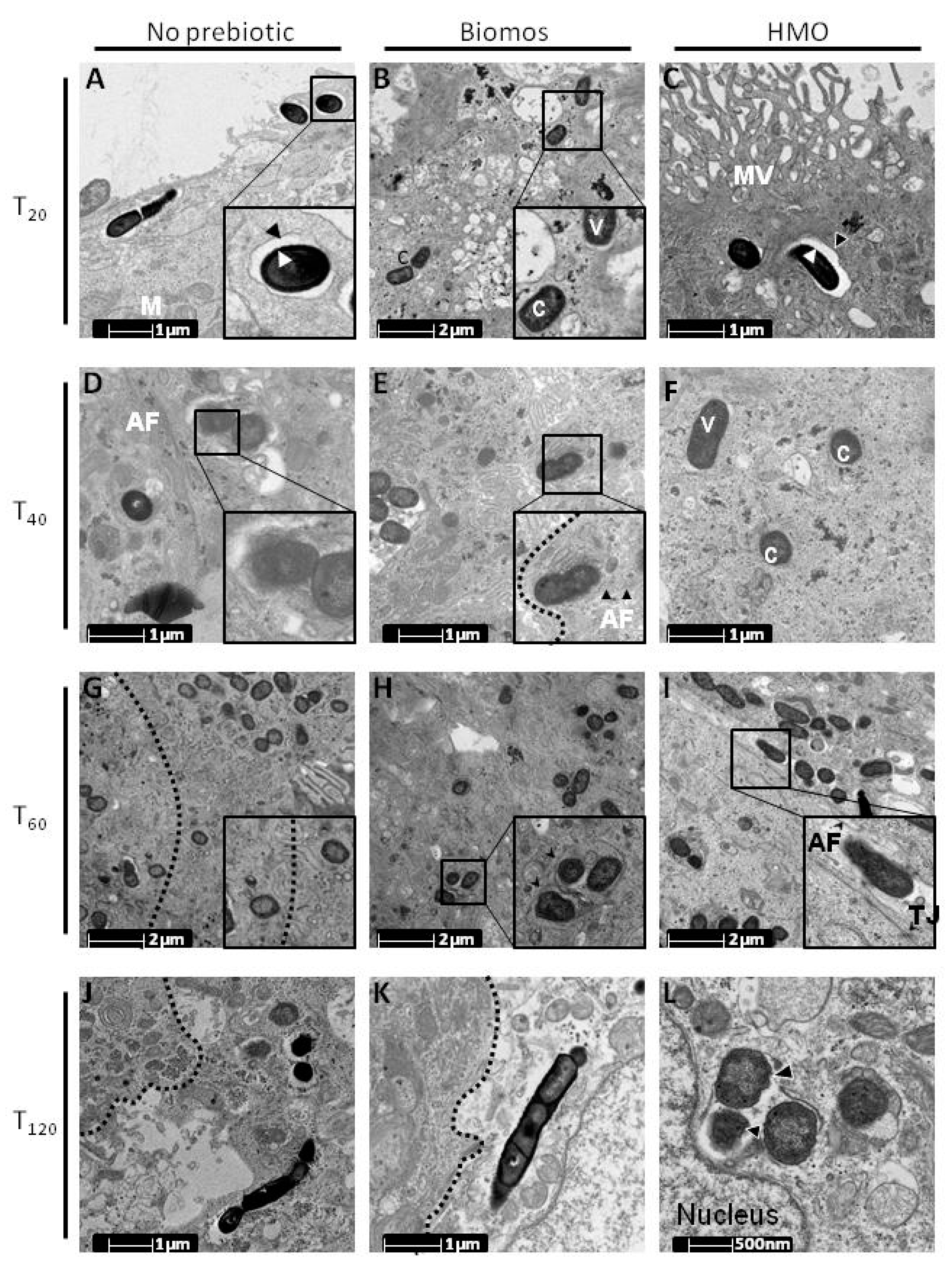
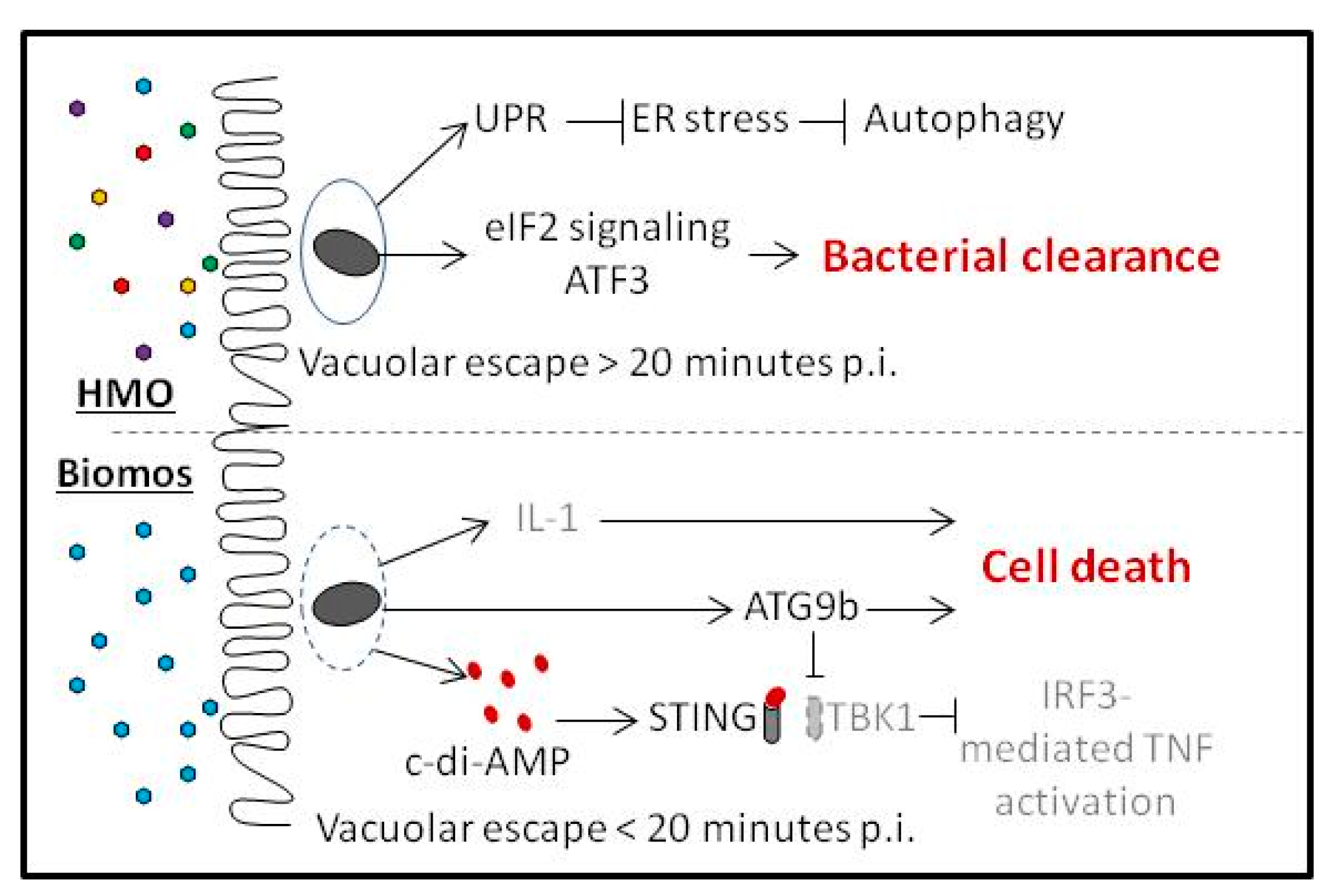
5. Biomos Expedites L. monocytogenes Vacuolar Escape
6. HMO Pretreatment Potentiates Host Cellular Intrinsic Protective Response during L. monocytogenes Infection
7. Discussion
8. Effect of Prebiotics on L. monocytogenes Host Association and Gene Expression
9. Prebiotic-Driven L. monocytogenes Vacuolar Escape
10. Host Cell Intrinsic Responses against L. monocytogenes Invasion
11. Materials and Methods
11.1. Oligosaccharides
11.2. Bacterial Strains and Growth Conditions
11.3. Human Cell Line and Growth Conditions
11.4. In Vitro Colonic Cell Infection Assays
11.5. RNA Extraction
11.6. RNAseq Library Preparation
11.7. Statistical Analysis for Differential Gene Expression
11.8. Gene Expression Pathway Analysis
11.9. Transmission Electron Microscopy
12. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Vital signs: Listeria illnesses, deaths, and outbreaks—United states, 2009–2011. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 448–452. [Google Scholar]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the united states—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Pizarro-Cerda, J.; Lecuit, M. Invasion of mammalian cells by listeria monocytogenes: Functional mimicry to subvert cellular functions. Trends Cell Biol. 2003, 13, 23–31. [Google Scholar] [CrossRef]
- Rogers, H.W.; Callery, M.P.; Deck, B.; Unanue, E.R. Listeria monocytogenes induces apoptosis of infected hepatocytes. J. Immunol. 1996, 156, 679–684. [Google Scholar] [PubMed]
- Valenti, P.; Greco, R.; Pitari, G.; Rossi, P.; Ajello, M.; Melino, G.; Antonini, G. Apoptosis of caco-2 intestinal cells invaded by listeria monocytogenes: Protective effect of lactoferrin. Exp. Cell Res. 1999, 250, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Casey, A.; Fox, E.M.; Schmitz-Esser, S.; Coffey, A.; McAuliffe, O.; Jordan, K. Transcriptome analysis of listeria monocytogenes exposed to biocide stress reveals a multi-system response involving cell wall synthesis, sugar uptake, and motility. Front. Microbiol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severino, P.; Dussurget, O.; Vencio, R.Z.; Dumas, E.; Garrido, P.; Padilla, G.; Piveteau, P.; Lemaitre, J.P.; Kunst, F.; Glaser, P.; et al. Comparative transcriptome analysis of listeria monocytogenes strains of the two major lineages reveals differences in virulence, cell wall, and stress response. Appl. Environ. Microbiol. 2007, 73, 6078–6088. [Google Scholar] [CrossRef] [PubMed]
- Milohanic, E.; Glaser, P.; Coppee, J.Y.; Frangeul, L.; Vega, Y.; Vazquez-Boland, J.A.; Kunst, F.; Cossart, P.; Buchrieser, C. Transcriptome analysis of listeria monocytogenes identifies three groups of genes differently regulated by prfa. Mol. Microbiol. 2003, 47, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Bahnan, W.; Wiley, D.J.; Barber, G.; Fields, K.A.; Schesser, K. Eukaryotic initiation factor 2 (eif2) signaling regulates proinflammatory cytokine expression and bacterial invasion. J. Biol. Chem. 2012, 287, 28738–28744. [Google Scholar] [CrossRef] [PubMed]
- Pillich, H.; Loose, M.; Zimmer, K.P.; Chakraborty, T. Activation of the unfolded protein response by listeria monocytogenes. Cell. Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Heinrichs, A.J.; Jones, C.M.; Heinrichs, B.S. Effects of mannan oligosaccharide or antibiotics in neonatal diets on health and growth of dairy calves. J. Dairy Sci. 2003, 86, 4064–4069. [Google Scholar] [CrossRef]
- Jones, G.H.; Ballou, C.E. Studies on the structure of yeast mannan. I. Purification and some properties of an alpha-mannosidase from an arthrobacter species. J. Biol. Chem. 1969, 244, 1043–1051. [Google Scholar] [PubMed]
- Coppa, G.V.; Pierani, P.; Zampini, L.; Bruni, S.; Carloni, I.; Gabrielli, O. Characterization of oligosaccharides in milk and feces of breast-fed infants by high-performance anion-exchange chromatography. Adv. Exp. Med. Biol. 2001, 501, 307–314. [Google Scholar] [PubMed]
- Ward, R.E.; Ninonuevo, M.; Mills, D.A.; Lebrilla, C.B.; German, J.B. In vitro fermentability of human milk oligosaccharides by several strains of bifidobacteria. Mol. Nutr. Food Res. 2007, 51, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Coppa, G.V.; Gabrielli, O.; Pierani, P.; Catassi, C.; Carlucci, A.; Giorgi, P.L. Changes in carbohydrate composition in human milk over 4 months of lactation. Pediatrics 1993, 91, 637–641. [Google Scholar] [PubMed]
- Coppa, G.V.; Zampini, L.; Galeazzi, T.; Facinelli, B.; Ferrante, L.; Capretti, R.; Orazio, G. Human milk oligosaccharides inhibit the adhesion to caco-2 cells of diarrheal pathogens: Escherichia coli, vibrio cholerae, and salmonella fyris. Pediatr. Res. 2006, 59, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Barboza, M.; Sonnenburg, E.D.; Pudlo, N.; Martens, E.C.; Desai, P.; Lebrilla, C.B.; Weimer, B.C.; Mills, D.A.; German, J.B.; et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 2011, 10, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; McCartney, A.L.; Rastall, R.A. Prebiotics and resistance to gastrointestinal infections. Br. J. Nutr. 2005, 93 (Suppl. 1), S31–S34. [Google Scholar] [CrossRef] [PubMed]
- Newburg, D.S.; Ruiz-Palacios, G.M.; Morrow, A.L. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 2005, 25, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, S.; Ridet, J.L.; Kusy, N.; Gao, H.; Crevoisier, F.; Guinchard, S.; Kochhar, S.; Sigrist, H.; Sprenger, N. Glycoprofiling with micro-arrays of glycoconjugates and lectins. Glycobiology 2005, 15, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Arabyan, N.; Park, D.; Foutouhi, S.; Weis, A.M.; Huang, B.C.; Williams, C.C.; Desai, P.; Shah, J.; Jeannotte, R.; Kong, N.; et al. Salmonella degrades the host glycocalyx leading to altered infection and glycan remodeling. Sci. Rep. 2016, 6, 29525. [Google Scholar] [CrossRef] [PubMed]
- Faith, N.; Kathariou, S.; Cheng, Y.; Promadej, N.; Neudeck, B.L.; Zhang, Q.; Luchansky, J.; Czuprynski, C. The role of l. Monocytogenes serotype 4b gtca in gastrointestinal listeriosis in a/j mice. Foodborne Pathog. Dis. 2009, 6, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Eugster, M.R.; Haug, M.C.; Huwiler, S.G.; Loessner, M.J. The cell wall binding domain of listeria bacteriophage endolysin plyp35 recognizes terminal glcnac residues in cell wall teichoic acid. Mol. Microbiol. 2011, 81, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Maga, E.A.; Desai, P.T.; Weimer, B.C.; Dao, N.; Kultz, D.; Murray, J.D. Consumption of lysozyme-rich milk can alter microbial fecal populations. Appl. Environ. Microbiol. 2012, 78, 6153–6160. [Google Scholar] [CrossRef] [PubMed]
- Barboza, M.; Pinzon, J.; Wickramasinghe, S.; Froehlich, J.W.; Moeller, I.; Smilowitz, J.T.; Ruhaak, L.R.; Huang, J.; Lonnerdal, B.; German, J.B.; et al. Glycosylation of human milk lactoferrin exhibits dynamic changes during early lactation enhancing its role in pathogenic bacteria-host interactions. Mol. Cell. Proteom. MCP 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Maga, E.A.; Weimer, B.C.; Murray, J.D. Dissecting the role of milk components on gut microbiota composition. Gut Microbes 2013, 4, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Liwosz, A.; Lei, T.; Kukuruzinska, M.A. N-glycosylation affects the molecular organization and stability of e-cadherin junctions. J. Biol. Chem. 2006, 281, 23138–23149. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R.M.; Cossart, P. E-cadherin is the receptor for internalin, a surface protein required for entry of l-monocytogenes into epithelial cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef]
- Aigal, S.; Claudinon, J.; Romer, W. Plasma membrane reorganization: A glycolipid gateway for microbes. Biochim. Biophys. Acta 2015, 1853, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Bohle, L.A.; Faergestad, E.M.; Veiseth-Kent, E.; Steinmoen, H.; Nes, I.F.; Eijsink, V.G.H.; Mathiesen, G. Identification of proteins related to the stress response in enterococcus faecalis v583 caused by bovine bile. Proteome Sci. 2010, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Barboza, M.; Pinzon, J.; German, J.B.; Weimer, B.C.; Lebrilla, C.B. N-glycans from human milk lactoferrin reduce adhesion and/or invasion of caco-2 cells by enteropathogenic bacteria. Glycobiology 2010, 20, 1534. [Google Scholar]
- Lee, J.H.; Karamychev, V.N.; Kozyavkin, S.A.; Mills, D.; Pavlov, A.R.; Pavlova, N.V.; Polouchine, N.N.; Richardson, P.M.; Shakhova, V.V.; Slesarev, A.I.; et al. Comparative genomic analysis of the gut bacterium bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genom. 2008, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad conservation of milk utilization genes in bifidobacterium longum subsp. Infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef] [PubMed]
- Comstock, S.S.; Li, M.; Wang, M.; Monaco, M.H.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Donovan, S.M. Dietary human milk oligosaccharides but not prebiotic oligosaccharides increase circulating natural killer cell and mesenteric lymph node memory T cell populations in noninfected and rotavirus-infected neonatal piglets. J. Nutr. 2017, 147, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Morrow, A.L.; Ruiz-Palacios, G.M.; Jiang, X.; Newburg, D.S. Human-milk glycans that inhibit pathogen binding protect breast-feeding infants against infectious diarrhea. J. Nutr. 2005, 135, 1304–1307. [Google Scholar] [PubMed]
- Slavin, J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex n-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Iji, P.A.; Saki, A.A.; Tivey, D.R. Intestinal structure and function of broiler chickens on diets supplemented with a mannan oligosaccharide. J. Sci. Food Agric. 2001, 81, 1186–1192. [Google Scholar] [CrossRef]
- Park, D.; Arabyan, N.; Williams, C.C.; Song, T.; Mitra, A.; Weimer, B.C.; Maverakis, E.; Lebrilla, C.B. Salmonella typhimurium enzymatically landscapes the host intestinal epithelial cell (iec) surface glycome to increase invasion. Mol. Cell. Proteom. 2016, 15, 3653–3664. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.H.; Mercer, L.P.; Flodin, N.W. General model for nutritional responses of higher organisms. Proc. Natl. Acad. Sci. USA 1975, 72, 4327–4331. [Google Scholar] [CrossRef] [PubMed]
- Abachin, E.; Poyart, C.; Pellegrini, E.; Milohanic, E.; Fiedler, F.; Berche, P.; Trieu-Cuot, P. Formation of D-alanyl-lipoteichoic acid is required for adhesion and virulence of listeria monocytogenes. Mol. Microbiol. 2002, 43, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Przybilla, K.; Stuhler, C.; Schauer, K.; Slaghuis, J.; Fuchs, T.M.; Goebel, W. Identification of listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J. Bacteriol. 2006, 188, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Lecuit, M. Interactions of listeria monocytogenes with mammalian cells during entry and actin-based movement: Bacterial factors, cellular ligands and signaling. EMBO J. 1998, 17, 3797–3806. [Google Scholar] [CrossRef]
- Witte, C.E.; Whiteley, A.T.; Burke, T.P.; Sauer, J.D.; Portnoy, D.A.; Woodward, J.J. Cyclic di-amp is critical for listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio 2013, 4, e00282-13. [Google Scholar] [CrossRef] [PubMed]
- Schwan, W.R.; Goebel, W. Host cell responses to listeria monocytogenes infection include differential transcription of host stress genes involved in signal transduction. Proc. Natl. Acad. Sci. USA 1994, 91, 6428–6432. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. C-di-amp secreted by intracellular listeria monocytogenes activates a host type i interferon response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Kaplan Zeevi, M.; Shafir, N.S.; Shaham, S.; Friedman, S.; Sigal, N.; Nir Paz, R.; Boneca, I.G.; Herskovits, A.A. Listeria monocytogenes multidrug resistance transporters and cyclic di-amp, which contribute to type i interferon induction, play a role in cell wall stress. J. Bacteriol. 2013, 195, 5250–5261. [Google Scholar] [CrossRef] [PubMed]
- Eiwegger, T.; Stahl, B.; Haidl, P.; Schmitt, J.; Boehm, G.; Dehlink, E.; Urbanek, R.; Szepfalusi, Z. Prebiotic oligosaccharides: In vitro evidence for gastrointestinal epithelial transfer and immunomodulatory properties. Pediatr. Allergy Immunol. UK 2010, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Kurakevich, E.; Hennet, T.; Hausmann, M.; Rogler, G.; Borsig, L. Milk oligosaccharide sialyl(alpha2,3)lactose activates intestinal cd11c+ cells through tlr4. Proc. Natl. Acad. Sci. USA 2013, 110, 17444–17449. [Google Scholar] [CrossRef] [PubMed]
- Jeurink, P.V.; van Esch, B.C.; Rijnierse, A.; Garssen, J.; Knippels, L.M. Mechanisms underlying immune effects of dietary oligosaccharides. Am. J. Clin. Nutr. 2013, 98, 572S–577S. [Google Scholar] [CrossRef] [PubMed]
- Van’t Land, B.; Schijf, M.; van Esch, B.C.; van Bergenhenegouwen, J.; Bastiaans, J.; Schouten, B.; Boon, L.; Garssen, J. Regulatory T-cells have a prominent role in the immune modulated vaccine response by specific oligosaccharides. Vaccine 2010, 28, 5711–5717. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Berthelsen, C.H.B.; Seemann, S.E.; Pan, X.Y.; Frederiksen, K.S.; Vilien, M.; Gorodkin, J.; Pociot, F. Transcriptomic landscape of lncrnas in inflammatory bowel disease. Genome Med. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumari, B.; Jain, P.; Das, S.; Ghosal, S.; Hazra, B.; Trivedi, A.C.; Basu, A.; Chakrabarti, J.; Vrati, S.; Banerjee, A. Dynamic changes in global micrornaome and transcriptome reveal complex mirna-mrna regulated host response to japanese encephalitis virus in microglial cells. Sci. Rep. 2016, 6, 20263. [Google Scholar] [CrossRef] [PubMed]
- Baddal, B.; Muzzi, A.; Censini, S.; Calogero, R.A.; Torricelli, G.; Guidotti, S.; Taddei, A.R.; Covacci, A.; Pizza, M.; Rappuoli, R.; et al. Dual RNA-seq of nontypeable haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. mBio 2015, 6, e01765-15. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Sandoval, F.; Poirier, M.; Scott, M.S. The emerging landscape of small nucleolar rnas in cell biology. Wiley Interdiscip. Rev. RNA 2015, 6, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Bartschat, S.; Tafer, H.; Stadler, P.F.; Hertel, J. Matching of soulmates: Coevolution of snornas and their targets. Mol. Biol. Evol. 2014, 31, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Lambeau, G. Emerging roles of secreted phospholipase a(2) enzymes: An update. Biochimie 2013, 95, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Statt, S.; Ruan, J.W.; Huang, C.T.; Wu, R.; Kao, C.Y. Lipidome and transcriptome profiling of pneumolysin intoxication identifies networks involved in statin-conferred protection of airway epithelial cells. Sci. Rep. 2015, 5, 10624. [Google Scholar] [CrossRef] [PubMed]
- Maraviglia, B.; Herring, F.G.; Weeks, G.; Godin, D.V. Detection of erythrocyte-membrane structural abnormalities in lecithin—Cholesterol acyltransferase deficiency using a spin label approach. J. Supramol. Struct. 1979, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J.J. Sting specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsdna-driven dynamic translocation of sting and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.R.; Barth, A.I.; Marquis, H.; de Hostos, E.L.; Nelson, W.J.; Theriot, J.A. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. Mol. Biol. Cell 1999, 146, 1333–1350. [Google Scholar]
- Nguyen, C.T.; Luong, T.T.; Lee, S.; Kim, G.L.; Pyo, S.; Rhee, D.K. ATF3 provides protection from staphylococcus aureus and listeria monocytogenes infections. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Strum, J.S.; Aldredge, D.; Barile, D.; Lebrilla, C.B. Coupling flash liquid chromatography with mass spectrometry for enrichment and isolation of milk oligosaccharides for functional studies. Anal. Biochem. 2012, 424, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, E.A. Measurement of invasion by gentamicin resistance. Methods Enzymol. 1994, 236, 405–420. [Google Scholar] [PubMed]
- Shah, J.; Desai, P.T.; Weimer, B.C. Genetic mechanisms underlying the pathogenicity of cold-stressed salmonella enterica serovar typhimurium in cultured intestinal epithelial cells. Appl. Environ. Microbiol. 2014, 80, 6943–6953. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Desai, P.T.; Chen, D.; Stevens, J.R.; Weimer, B.C. Preadaptation to cold stress in salmonella enterica serovar typhimurium increases survival during subsequent acid stress exposure. Appl. Environ. Microbiol. 2013, 79, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian micrornas predominantly act to decrease target mrna levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, N.; Ng, W.; Lee, V.; Weimer, B.C. Production and Analysis of High Molecular Weight Genomic DNA for NGS Pipelines Using Agilent DNA Extraction Kit (p/n 200600); Agilent Technologies: Santa Clara, CA, USA, 2013. [Google Scholar]
- Kol, A.; Foutouhi, S.; Walker, N.J.; Kong, N.T.; Weimer, B.C.; Borjesson, D.L. Gastrointestinal microbes interact with canine adipose-derived mesenchymal stem cells in vitro and enhance immunomodulatory functions. Stem Cells Dev. 2014, 23, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The metacyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Latendresse, M.; Paley, S.M.; Krummenacker, M.; Ong, Q.D.; Billington, R.; Kothari, A.; Weaver, D.; Lee, T.; Subhraveti, P.; et al. Pathway tools version 19.0 update: Software for pathway/genome informatics and systems biology. Brief. Bioinform. 2016, 17, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or depletion of a go category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Pathways | p-Value | Genes Matched |
|---|---|---|---|
| Redox | Cobalamin Biosynthesis | 0.003 | cobD, lmo1164, lmo1181 |
| Cofactors, Prosthetic Groups, Electron Carriers Biosynthesis | 0.008 | nadE, nadD, hemE, menE, hemA, cobD, serC, panB, thyA, lmo2749, lmo2101, lmo0938, lmo1930, lmo1164, lmo1181 | |
| Vitamins Biosynthesis | 0.012 | cobD, serC, panB, thyA, lmo2749, lmo2101, lmo1164, lmo1181 | |
| Adenosylcobalamin Biosynthesis | 0.008 | lmo1164, lmo1181 | |
| Adenosylcobalamin salvage from cobalamin | 0.008 | ||
| Biotin-carboxyl carrier protein assembly | 0.022 | accD, lmo1357 | |
| Vitamin B6 Biosynthesis | 0.022 | serC, lmo2101 | |
| Amino acid metabolism | Amino Acids Degradation | 0.006 | gltD, aspB, lmo2836, lmo2363, lmo0383, lmo1182, lmo2101, lmo2749, lmo1915 |
| Proteinogenic Amino Acids Degradation | 0.006 | ||
| L-aspartate Degradation | 0.022 | aspB, lmo1915 | |
| Aspartate degradation II | 0.022 | ||
| Aspartate superpathway | 0.047 | nadE, nadD, aspB, lmo1436, lmo1437 | |
| Pyrimidine deoxyribonucleosides salvage | 0.022 | thyA, lmo1463 | |
| Pyrimidine deoxyribonucleotides de novo biosynthesis II | 0.042 | thyA, lmo0279 | |
| L-threonine Biosynthesis | 0.039 | aspB, lmo1436, lmo1437 | |
| Threonine biosynthesis | 0.039 | ||
| Energy metabolism | Degradation/Utilization/Assimilation | 0.026 | gltD, aspB, lmo0347, lmo2696, lmo1057, lmo2648, lmo2836, lmo1463, lmo0877, lmo1182, lmo0383, lmo2101, lmo2749, lmo0372, lmo0917, lmo2771, lmo2840, lmo1915, lmo2362, lmo2095, lmo2835 |
| Secondary metabolite degradation | Taxiphyllin bioactivation | 0.028 | lmo0372, lmo0917, lmo2771 |
| Nitrogen Containing Glucosides Degradation | 0.028 | ||
| Cyanogenic Glucosides Degradation | 0.028 | ||
| Nitrogen Containing Secondary Compounds Degradation | 0.039 |
| Gene Function | Gene Name | Ensembl ID | Lm/+HMO | Lm/+Biomos | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Log2 FC | LogCPM | p-Value | FDR | Log2 FC | LogCPM | p-Value | FDR | |||
| RNA-associated proteins, small RNAs and pseudo-genes | SNORA53/ACA53 | ENSG00000212443 | −3.347 | 1.672 | 3.25 × 10−8 | 0.00021 | ns | -- | -- | -- |
| RPPH1 | ENSG00000277209 | −1.420 | 4.499 | 2.5 × 10−5 | 0.04977 | −3.598 | 4.651 | 2.5 × 10−11 | 2.8 × 10−9 | |
| RPPH1 | ENSG00000259001 | −1.417 | 4.502 | 2.5 × 10−5 | 0.04977 | −3.584 | 4.653 | 1.9 × 10−11 | 2.3 × 10−9 | |
| AC010761.8 | ENSG00000264577 | −1.302 | 3.546 | 7.6 × 10−7 | 0.00374 | −3.269 | 3.650 | 1.6 × 10−18 | 2.1 × 10−15 | |
| RN7SL5P | ENSG00000278249 | −1.553 | 1.421 | 2.5 × 10−6 | 0.00605 | −1.797 | 1.729 | 2.3 × 10−5 | 2.0 × 10−4 | |
| SCARNA2 | ENSG00000270066 | −1.553 | 1.421 | 2.5 × 10−6 | 0.00605 | −1.797 | 1.729 | 2.3 × 10−5 | 2.0 × 10−4 | |
| RN7SL5P | ENSG00000265735 | −1.243 | 2.999 | 1.5 × 10−6 | 0.00503 | −1.658 | 3.278 | 3.0 × 10−5 | 2.4 × 10−4 | |
| SnoU2_19 | ENSG00000275146 | −1.138 | 1.452 | 1.2 × 10−6 | 0.00451 | −1.340 | 1.747 | 1.1 × 10−3 | 4.3 × 10−3 | |
| RMRP | ENSG00000277027 | −1.316 | 3.178 | 2.6 × 10−8 | 0.00021 | −1.182 | 3.614 | 1.1 × 10−2 | 2.9 × 10−2 | |
| RMRP | ENSG00000269900 | −1.315 | 3.178 | 2.8 × 10−8 | 0.00021 | −1.182 | 3.614 | 1.1 × 10−2 | 2.9 × 10−2 | |
| AC084809.2 | ENSG00000226377 | ns | -- | -- | -- | −3.743 | 2.709 | 5.4 × 10−20 | 9.3 × 10−17 | |
| RPL23A | ENSG00000198242 | ns | -- | -- | -- | −3.530 | 6.238 | 3.2 × 10−25 | 1.4 × 10−21 | |
| RP11-3P17.5 | ENSG00000269888 | ns | -- | -- | -- | −3.511 | 1.359 | 3.6 × 10−21 | 8.9 × 10−18 | |
| RPL23AP42 | ENSG00000234851 | ns | -- | -- | -- | −3.511 | 1.359 | 3.6 × 10−21 | 8.9 × 10−18 | |
| RN7SL2 | ENSG00000274012 | ns | -- | -- | -- | −3.330 | 7.607 | 5.1 × 10−11 | 4.9 × 10−9 | |
| AL627171.2 | ENSG00000282885 | ns | -- | -- | -- | −3.317 | 7.609 | 3.2 × 10−11 | 3.4 × 10−9 | |
| AC010761.1 | ENSG00000264577 | ns | -- | -- | -- | −3.269 | 3.650 | 1.6 × 10−18 | 2.1 × 10−15 | |
| RN7SL255P | ENSG00000239808 | ns | -- | -- | -- | −2.975 | 0.630 | 5.3 × 10−8 | 1.4 × 10−6 | |
| AC121158.1 | ENSG00000231335 | ns | -- | -- | -- | −2.707 | 2.933 | 4.6 × 10−13 | 9.6 × 10−11 | |
| MALAT1 | ENSG00000278217 | ns | -- | -- | -- | −2.701 | 3.849 | 2.05 × 10−13 | 4.8 × 10−11 | |
| C11orf98 | ENSG00000278615 | ns | -- | -- | -- | −2.683 | 1.110 | 5.45 × 10−17 | 4.5 × 10−14 | |
| CCDC74A | ENSG00000163040 | ns | -- | -- | -- | 2.013 | 0.254 | 2.40 × 10−8 | 7.2 × 10−7 | |
| AC022966.2 | ENSG00000267601 | ns | -- | -- | -- | 2.057 | 0.132 | 3.68 × 10−7 | 6.7 × 10−6 | |
| NRA5A1 | ENSG00000136931 | ns | -- | -- | -- | 2.078 | 2.664 | 2.93 × 10−18 | 3.5 × 10−15 | |
| RP11-432M8.5 | ENSG00000249329 | ns | -- | -- | -- | 2.079 | 0.360 | 3.51 × 10−11 | 3.6 × 10−9 | |
| AC113208.3 | ENSG00000260660 | ns | -- | -- | -- | 2.111 | 0.784 | 5.66 × 10−9 | 2.2 × 10−9 | |
| AC017104.6 | ENSG00000224376 | ns | -- | -- | -- | 2.122 | −0.211 | 4.61 × 10−10 | 2.9 × 10−8 | |
| KRT16P2 | ENSG00000227300 | ns | -- | -- | -- | 2.142 | −0.021 | 2.7 × 10−8 | 7.8 × 10−7 | |
| PPP6R2P1 | ENSG00000233442 | ns | -- | -- | -- | 2.148 | −0.062 | 9.5 × 10−9 | 3.4 × 10−7 | |
| CA15P1 | ENSG00000241527 | ns | -- | -- | -- | 2.172 | −0.143 | 4.3 × 10−6 | 5.0 × 10−5 | |
| FAM90A22P | ENSG00000215365 | ns | -- | -- | -- | 2.189 | −0.027 | 1.8 × 10−9 | 8.7 × 10−8 | |
| BTBD17 | ENSG00000204347 | ns | -- | -- | -- | 2.215 | 0.772 | 1.2 × 10−9 | 6.5 × 10−8 | |
| RP11-26H16.4 | ENSG00000283234 | ns | -- | -- | -- | 2.309 | 0.035 | 1.6 × 10−11 | 2.0 × 10−9 | |
| RP11-331F4.5 | ENSG00000280152 | ns | -- | -- | -- | 2.348 | 0.207 | 3.5 × 10−10 | 2.3 × 10−8 | |
| SLC25A34 | ENSG00000162461 | ns | -- | -- | -- | 2.904 | −0.043 | 4.9 × 10−12 | 7.5 × 10−10 | |
| Tran-scriptional regulators | ZBED6 | ENSG00000257315 | ns | -- | -- | -- | −2.625 | 2.892 | 5.7 × 10−15 | 2.6 × 10−12 |
| ZNF703 | ENSG00000183779 | ns | -- | -- | -- | 2.680 | 2.689 | 1.3 × 10−18 | 1.9 × 10−15 | |
| HO × D10 | ENSG00000128710 | ns | -- | -- | -- | 2.041 | 0.792 | 8.3 × 10−11 | 7.4 × 10−9 | |
| Cell cycle | NAMPTP1 | ENSG00000229644 | ns | -- | -- | -- | −3.272 | 0.963 | 1.5 × 10−26 | 8.6 × 10−23 |
| GADD45A | ENSG00000116717 | ns | -- | -- | -- | −2.684 | 2.171 | 3.0 × 10−16 | 2.0 × 10−13 | |
| Inflammation | TNFAIP3 | ENSG00000118503 | ns | -- | -- | -- | −3.054 | 4.040 | 7.2 × 10−21 | 1.6 × 10−17 |
| JUN | ENSG00000177606 | ns | -- | -- | -- | −3.436 | 3.904 | 3.9 × 10−33 | 4.4 × 10−29 | |
| FOS | ENSG00000170345 | ns | -- | -- | -- | −2.871 | 3.398 | 9.4 × 10−16 | 5.4 × 10−13 | |
| Endoplasmic reticulum stress | HSPA1A | ENSG00000204389 | ns | -- | -- | -- | −2.883 | 3.702 | 1.8 × 10−34 | 4.2 × 10−30 |
| HSPA1B | ENSG00000204388 | ns | -- | -- | -- | −2.749 | 3.423 | 6.5 × 10−30 | 4.9 × 10−26 | |
| PSMD10 | ENSG00000101843 | ns | -- | -- | -- | −2.749 | 1.833 | 1.0 × 10−22 | 3.9 × 10−19 | |
| Cell structure | SPTBN5 | ENSG00000137877 | ns | -- | -- | -- | 2.017 | 3.286 | 3.7 × 10−16 | 2.4 × 10−13 |
| MYL3 | ENSG00000160808 | ns | -- | -- | -- | 2.125 | 2.038 | 4.5 × 10−15 | 2.1 × 10−12 | |
| Receptors | GRASP | ENSG00000161835 | ns | -- | -- | -- | 2.068 | 1.589 | 1.1 × 10−13 | 2.8 × 10−11 |
| TMEM82 | ENSG00000162460 | ns | -- | -- | -- | 2.091 | 0.168 | 1.6 × 10−8 | 5.1 × 10−7 | |
| CHRND | ENSG00000135902 | ns | -- | -- | -- | 2.130 | 0.405 | 1.9 × 10−10 | 1.4 × 10−8 | |
| UCHL1 | ENSG00000154277 | ns | -- | -- | -- | 2.165 | 3.558 | 7.8 × 10−19 | 1.2 × 10−15 | |
| GPR142 | ENSG00000257008 | ns | -- | -- | -- | 2.211 | 0.751 | 2.1 × 10−11 | 2.4 × 10−9 | |
| GFRA4 | ENSG00000125861 | ns | -- | -- | -- | 2.218 | 0.069 | 5.2 × 10−9 | 2.1 × 10−7 | |
| CABP7 | ENSG00000100314 | ns | -- | -- | -- | 2.285 | 1.519 | 3.1 × 10−10 | 2.1 × 10−8 | |
| Gene Function | Gene Name | Ensembl ID | HMO/+Lm | Biomos/+Lm | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Log2 FC | LogCPM | p-Value | FDR | Log2 FC | LogCPM | p-Value | FDR | |||
| ER stress and protein folding | YOD1 | ENSG00000180667 | 1.927 | 1.527 | 1.3 × 10−5 | 9.1 × 10−3 | −2.505 | 1.413 | 1.1 × 10−14 | 1.1 × 10−11 |
| HSPA1B | ENSG00000204388 | 3.146 | 3.801 | 1.8 × 10−9 | 1.1 × 10−5 | −3.009 | 3.528 | 6.2 × 10−30 | 3.5 × 10−26 | |
| HSPA1A | ENSG00000204389 | 3.228 | 4.070 | 1.1 × 10−10 | 1.2 × 10−6 | −3.145 | 3.813 | 1.1 × 10−32 | 1.6 × 10−28 | |
| HSPH1 | ENSG00000120694 | 2.062 | 4.095 | 3.3 × 10−5 | 1.5 × 10−2 | −1.579 | 3.787 | 4.6 × 10−12 | 1.6 × 10−9 | |
| INSIG1 | ENSG00000186480 | 2.432 | 4.979 | 2.6 × 10−6 | 4.3 × 10−3 | −1.480 | 4.788 | 5.7 × 10−8 | 3.2 × 10−6 | |
| PSMD10 | ENSG00000101843 | 1.912 | 1.654 | 2.6 × 10−6 | 1.3 × 10−2 | −2.668 | 1.579 | 2.6 × 10−12 | 1.0 × 10−9 | |
| Inflammation | TNFAIP3 | ENSG00000118503 | ns | -- | -- | -- | −2.897 | 3.777 | 1.4 × 10−32 | 1.6 × 10−28 |
| NFKB1A | ENSG00000100906 | ns | -- | -- | -- | −2.591 | 2.723 | 4.0 × 10−22 | 1.5 × 10−18 | |
| JUN | ENSG00000177606 | ns | -- | -- | -- | −3.407 | 3.751 | 1.7 × 10−31 | 1.3 × 10−27 | |
| Cell cycle | GADD45A | ENSG00000116717 | 2.083 | 2.346 | 4.0 × 10−5 | 1.7 × 10−2 | −2.878 | 2.215 | 6.8 × 10−25 | 3.1 × 10−21 |
| TXNIP | ENSG00000265972 | 2.645 | 5.494 | 5.5 × 10−5 | 1.8 × 10−2 | −2.066 | 5.187 | 4.4 × 10−12 | 1.5 × 10−9 | |
| NAMPT | ENSG00000105835 | 2.325 | 5.180 | 2.3 × 10−5 | 1.2 × 10−2 | −2.363 | 5.024 | 1.3 × 10−12 | 5.8 × 10−10 | |
| NAMPTP1 | ENSG00000229644 | 2.563 | 0.994 | 6.5 × 10−6 | 7.4 × 10−3 | −3.402 | 0.934 | 5.5 × 10−22 | 1.8 × 10−18 | |
| Miscellaneous functions | TIPARP | ENSG00000163659 | 2.557 | 5.719 | 3.4 × 10−6 | 5.3 × 10−3 | −1.055 | 5.777 | 1.2 × 10−5 | 1.9 × 10−4 |
| CD164 | ENSG00000135535 | 2.280 | 5.575 | 4.8 × 10−5 | 1.7 × 10−2 | −1.970 | 5.610 | 2.7 × 10−10 | 4.5 × 10−8 | |
| GGCT | ENSG00000006625 | 2.154 | 1.467 | 1.1 × 10−5 | 9.1 × 10−3 | −2.651 | 1.532 | 2.2 × 10−10 | 3.9 × 10−8 | |
| Transcriptional regulator | FOS | ENSG00000170345 | 2.520 | 3.030 | 4.6 × 10−8 | 1.5 × 10−4 | −2.468 | 2.890 | 2.4 × 10−13 | 1.4 × 10−10 |
| ZNF703 | ENSG00000183779 | −2.001 | 2.473 | 2.9 × 10−4 | 3.8 × 10−2 | 2.141 | 2.674 | 4.6 × 10−16 | 5.7 × 10−13 | |
| ZBED6 | ENSG00000257315 | 2.622 | 2.535 | 5.6 × 10−6 | 7.4 × 10−3 | −2.181 | 2.362 | 7.9 × 10−11 | 1.7 × 10−8 | |
| KLF6 | ENSG00000067082 | 2.010 | 5.008 | 1.6 × 10−4 | 3.0 × 10−2 | −1.625 | 4.845 | 8.8 × 10−8 | 4.5 × 10−6 | |
| RNA⁻associated proteins, small RNAs and pseudogenes | HNRNPA1 | ENSG00000135486 | 2.397 | 5.602 | 5.1 × 10−5 | 1.7 × 10−2 | −2.433 | 5.567 | 8.5 × 10−17 | 1.2 × 10−13 |
| RPL23A | ENSG00000198242 | 2.198 | 5.435 | 2.3 × 10−8 | 8.7 × 10−5 | −3.133 | 5.742 | 4.1 × 10−21 | 1.2 × 10−17 | |
| RPPH1 | ENSG00000277209 | 1.596 | 3.351 | 1.9 × 10−6 | 3.6 × 10−3 | −2.905 | 3.885 | 1.7 × 10−8 | 1.3 × 10−6 | |
| MRPL36 | ENSG00000171421 | 2.083 | 1.352 | 1.4 × 10−9 | 1.1 × 10−5 | −2.502 | 1.916 | 4.0 × 10−15 | 4.5 × 10−12 | |
| RN7SL255P | ENSG00000239808 | 4.349 | 0.639 | 3.2 × 10−9 | 1.5 × 10−5 | −2.712 | 0.329 | 2.2 × 10−13 | 1.4 × 10−10 | |
| KRT17P4 | ENSG00000205312 | −1.575 | 0.132 | 4.3 × 10−4 | 4.3 × 10−2 | 2.258 | 0.067 | 8.1 × 10−11 | 1.8 × 10−8 | |
| RPL23AP42 | ENSG00000234851 | 1.831 | 0.804 | 1.3 × 10−5 | 9.1 × 10−3 | −3.126 | 0.824 | 7.1 × 10−14 | 5.2 × 10−11 | |
| AC084809.2 | ENSG00000226377 | 4.358 | 2.636 | 2.3 × 10−15 | 5.4 × 10−11 | −3.823 | 2.643 | 5.6 × 10−18 | 1.1 × 10−14 | |
| AC107072.2 | ENSG00000231335 | 2.105 | 2.386 | 4.4 × 10−5 | 1.7 × 10−2 | −1.749 | 1.998 | 4.6 × 10−11 | 1.2 × 10−8 | |
| CTD-202417.13 | ENSG00000246422 | 2.286 | 1.332 | 6.4 × 10−6 | 7.4 × 10−3 | −1.227 | 1.357 | 1.1 × 10−6 | 3.1 × 10−5 | |
| MACC1-AS1 | ENSG00000228598 | 2.466 | 1.806 | 6.7 × 10−7 | 1.8 × 10−3 | −2.101 | 1.410 | 5.2 × 10−13 | 2.8 × 10−10 | |
| CTD-3014M21.1 | ENSG00000279602 | 2.594 | 2.937 | 9.4 × 10−6 | 8.6 × 10−3 | −1.983 | 2.744 | 1.7 × 10−11 | 4.8 × 10−9 | |
| RPPH1 | ENSG00000259001 | 1.590 | 3.357 | 2.0 × 10−6 | 3.6 × 10−3 | −2.893 | 3.890 | 1.4 × 10−8 | 1.1 × 10−6 | |
| RP11-3P17.5 | ENSG00000269888 | 1.823 | 0.806 | 1.0 × 10−5 | 8.9 × 10−3 | −3.126 | 0.824 | 7.1 × 10−14 | 5.2 × 10−11 | |
| RPS13 | ENSG00000110700 | 1.784 | 5.309 | 4.2 × 10−5 | 1.7 × 10−2 | −2.705 | 5.665 | 1.5 × 10−13 | 10.0 × 10−11 | |
| AC010761.8 | ENSG00000264577 | 1.296 | 2.570 | 3.2 × 10−4 | 3.9 × 10−2 | −2.607 | 2.914 | 6.5 × 10−13 | 3.4 × 10−10 | |
| RN7SL2 | ENSG00000274012 | ns | -- | -- | -- | −2.892 | 7.104 | 3.8 × 10−8 | 2.4 × 10−6 | |
| RP11-596C23.6 | ENSG00000282885 | ns | -- | -- | -- | −2.880 | 7.107 | 2.6 × 10−8 | 1.8 × 10−6 | |
| DNASE1L3 | ENSG00000283148 | ns | -- | -- | -- | 2.226 | −0.175 | 6.2 × 10−11 | 1.5 × 10−8 | |
| RP11-321E2.8-001 | ENSG00000250055 | ns | -- | -- | -- | 2.400 | −0.013 | 4.9 × 10−13 | 2.7 × 10−10 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.; Reiter, T.; Huang, B.; Kong, N.; Weimer, B.C. Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection. Pathogens 2017, 6, 68. https://doi.org/10.3390/pathogens6040068
Chen P, Reiter T, Huang B, Kong N, Weimer BC. Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection. Pathogens. 2017; 6(4):68. https://doi.org/10.3390/pathogens6040068
Chicago/Turabian StyleChen, Poyin, Taylor Reiter, Bihua Huang, Nguyet Kong, and Bart C. Weimer. 2017. "Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection" Pathogens 6, no. 4: 68. https://doi.org/10.3390/pathogens6040068
APA StyleChen, P., Reiter, T., Huang, B., Kong, N., & Weimer, B. C. (2017). Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection. Pathogens, 6(4), 68. https://doi.org/10.3390/pathogens6040068