Vertical Transmission of Listeria monocytogenes: Probing the Balance between Protection from Pathogens and Fetal Tolerance

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structural and Physiological Comparison of Animal Models Used to Assess L. monocytogenes Vertical Transmission
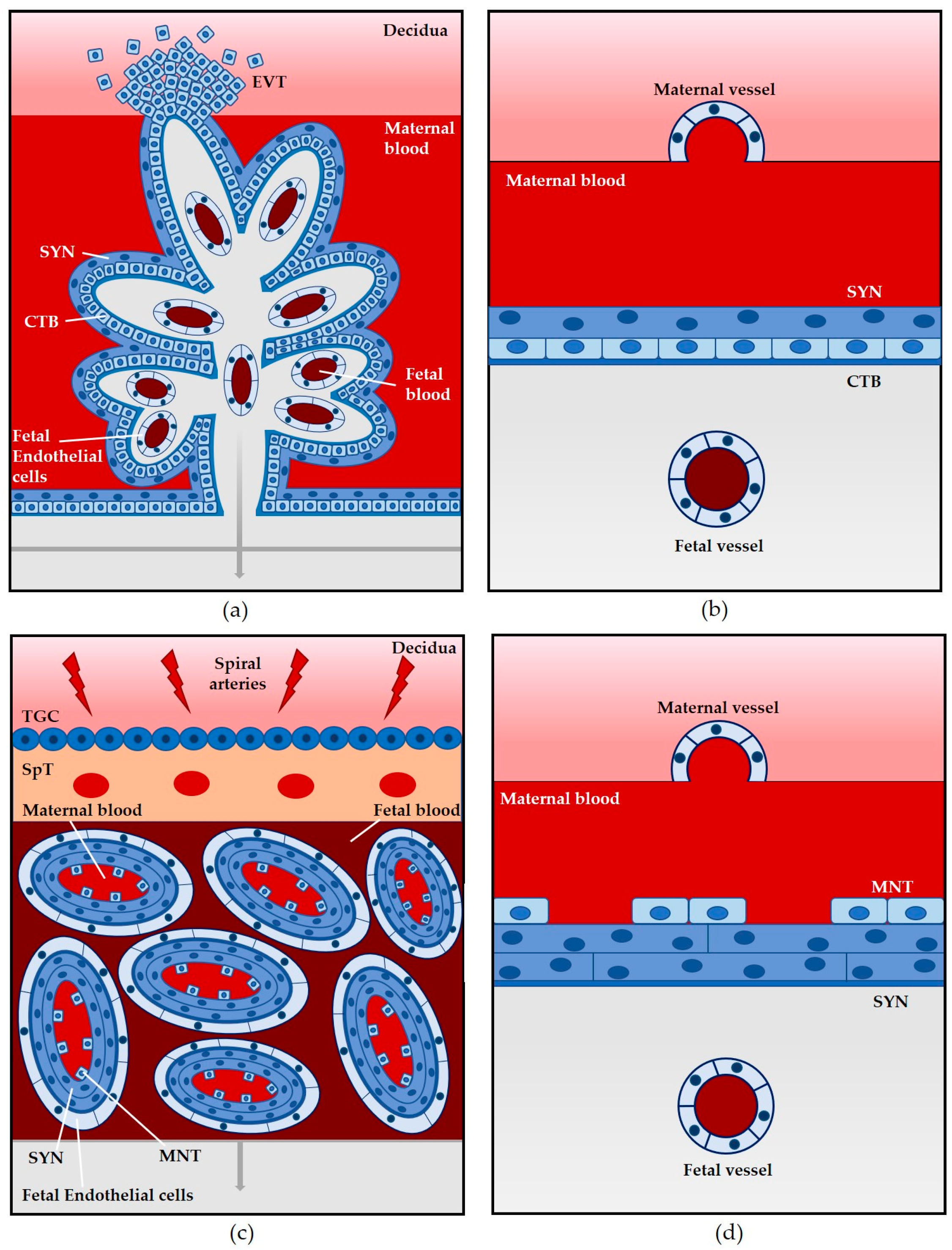
2.1. Mouse
2.2. Guinea Pig
2.3. Gerbil
2.4. Non-Human Primates
3. Lessons from Available Animal Models: Routes of Placental Entry and Bacterial Factors that Contribute to L. monocytogenes Vertical Transmission
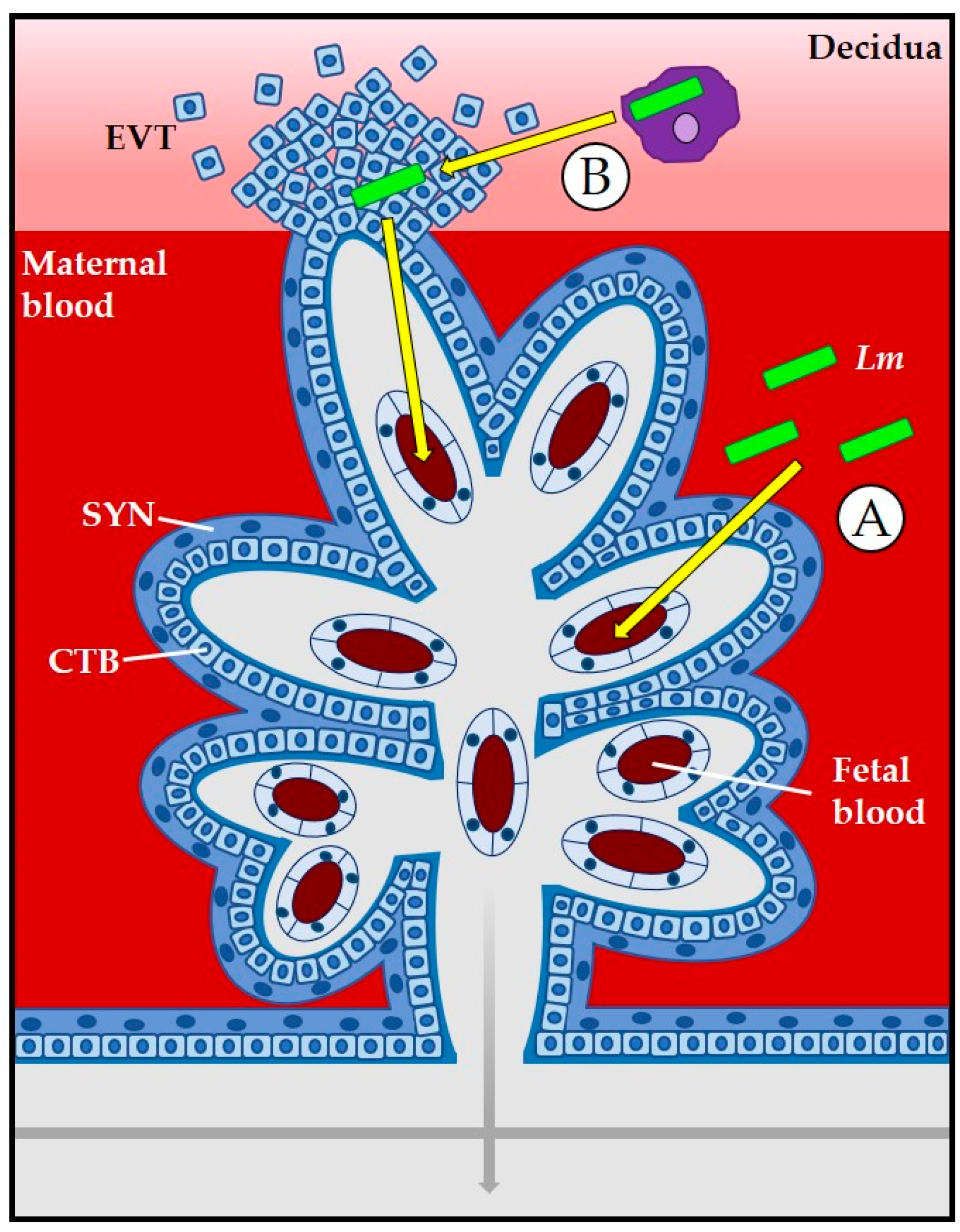
3.1. The Placenta as a Barrier to L. monocytogenes Infection
3.2. Role of Bacterial Internalin Proteins in Placental and Fetal Invasion
3.3. Role of LLO and Phospholipases
3.4. Role of ActA
4. Balancing Fetal Tolerance with Protection against Pathogens: Maternal Immune Responses to L. monocytogenes
4.1. Cells and Cell Signaling at the Maternofetal Interface
4.2. Maternal Immune Responses to L. monocytogenes during Pregnancy
5. Summary
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Robbins, J.R.; Bakardjiev, A.I. Pathogens and the placental fortress. Curr. Opin. Microbiol. 2012, 15, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Greub, G. Intracellular bacteria and adverse pregnancy outcomes. Clin. Microbiol. Infect. 2011, 17, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Dussurget, O.; Pizarro-Cerda, J.; Cossart, P. Molecular determinants of Listeria monocytogenes virulence. Ann. Rev. Microbial. 2004, 58, 587–610. [Google Scholar] [CrossRef] [PubMed]
- Freitag, N.E. From hot dogs to host cells: How the bacterial pathogen Listeria monocytogenes regulates virulence gene expression. Future Microbial. 2006, 1, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Hain, T.; Chatterjee, S.S.; Ghai, R.; Kuenne, C.T.; Billion, A.; Steinweg, C.; Domann, E.; Kärst, U.; Jänsch, L.; Wehland, J.; et al. Pathogenomics of Listeria spp. Int. J. Med. Microbiol. 2007, 297, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Freitag, N.E.; Boor, K.J. How the Bacterial Pathogen Listeria monocytogenes Mediates the Switch from Environmental Dr. Jekyll to Pathogenic Mr. Hyde. Infect. Immun. 2006, 74, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Port, G.C.; Miner, M.D.; Freitag, N.E. Listeria monocytogenes-from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 2009, 7, 623–628. [Google Scholar] [CrossRef]
- Gray, M.L.; Killinger, A.H. Listeria monocytogenes and listeric infections. Bacteriol. Rev. 1966, 30, 309–382. [Google Scholar] [PubMed]
- Seeliger, H.P.R.; Finger, H. Listeriosis; W.B. Saunders Co: Philadelphia, PA, USA, 1976; pp. 333–365. [Google Scholar]
- Seeliger, H.P. Listeriosis–history and actual developments. Infection 1988, 16 (Suppl. 2), S84. [Google Scholar] [CrossRef]
- Vázquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Domínguez-Bernal, G.; Goebel, W.; González-Zorn, B.; Wehland, J.; Kreft, J. Listeria Pathogenesis and Molecular Virulence Determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. (CDC) Multistate Outbreak of Listeriosis–United States, 1998. Morb. Mortal. Wkly. Rep. 1998, 47, 1085–1086. [Google Scholar]
- Centers for Disease Control and Prevention. (CDC) Update: Multistate Outbreak of Listeriosis—United States, 1998–1999. Morb. Mortal. Wkly. Rep. 1999, 47, 1117–1118. [Google Scholar]
- Centers for Disease Control and Prevention. (CDC) Preliminary FoodNet data on the incidence of infection with pathogens transmitted commony through food—Selected sites, United States, 2003. Morb. Mortal. Wkly. Rep. 2004, 53, 338–343. [Google Scholar]
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S.; Dunne, E.F.; Graves, L.; Wiedmann, M.; Patrick, M.; Hunter, S.; Salehi, E.; Mostashari, F.; Craig, A.; Mshar, P.; et al. Nationwide outbreak of listeriosis due to contaminated meat. Epidemiol. Infect. 2006, 134, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Drevets, D.A.; Bronze, M.S. Listeria monocytogenes: Epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol. Med. Microbiol. 2008, 53, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. (CDC) National Listeria Surveillance Annual Summary, 2013; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2015.
- Centers for Disease Control and Prevention. (CDC) Vital signs: Listeria illnesses, deaths, and outbreaks–United States, 2009–2011. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 448–452. [Google Scholar]
- Madjunkov, M.; Chaudhry, S.; Ito, S. Listeriosis during pregnancy. Arch. Gynecol. Obstet. 2017, 296, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.R.; Skrzypczynska, K.M.; Zeldovich, V.B.; Kapidzic, M.; Bakardjiev, A.I. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathog. 2010, 6, e1000732. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Bakardjiev, A.I.; Fisher, S.J. The placenta: Transcriptional, epigenetic, and physiological integration during development. J. Clin. Investig. 2010, 120, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Animal Models of Human Placentation–A Review. Placenta 2007, 28, S47. [Google Scholar] [CrossRef] [PubMed]
- Bakardjiev, A.I.; Stacy, B.A.; Fisher, S.J.; Portnoy, D.A. Listeriosis in the Pregnant Guinea Pig: A Model of Vertical Transmission. Infect. Immun. 2004, 72, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M. Human listeriosis and animal models. Microbes Infect. 2007, 9, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Le Monnier, A.; Join-Lambert, O.F.; Jaubert, F.; Berche, P.; Kayal, S. Invasion of the Placenta during Murine Listeriosis. Infect. Immun. 2006, 74, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.; Wiepz, G.J.; Schotzko, M.; Bondarenko, G.I.; Durning, M.; Simmons, H.A.; Mejia, A.; Faith, N.G.; Sampene, E.; Suresh, M.; et al. Acute Fetal Demise with First Trimester Maternal Infection Resulting from Listeria monocytogenes in a Nonhuman Primate Model. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Nikitas, G.; Ragon, M.; Cossart, P.; Dussurget, O.; Lecuit, M.; Le Monnier, A.; Grayo, S.; Babinet, C.; Disson, O.; Huillet, E.; et al. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 2008, 455, 1114–1118. [Google Scholar] [CrossRef]
- Moffett, A.; Loke, C. Immunology of placentation in eutherian mammals. Nat. Rev. Immunol. 2006, 6, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mège, R.; Cossart, P. E-Cadherin Is the Receptor for Internalin, a Surface Protein Required for Entry of L. monocytogenes into Epithelial Cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef]
- Fedor-Chaiken, M.; Cossart, P.; Dramsi, S.; Lecuit, M.; Gottardi, C.; Gumbiner, B. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999, 18, 3956–3963. [Google Scholar] [CrossRef]
- Tsai, Y.; Disson, O.; Bierne, H.; Lecuit, M. Murinization of Internalin Extends Its Receptor Repertoire, Altering Listeria monocytogenes Cell Tropism and Host Responses. PLoS Pathog. 2013, 9, e1003381. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Pasche, B.; Rochon, M.; Deppenmeier, S.; van den Heuvel, J.; Gruber, A.; Heinz, D.W.; Lengeling, A.; Schubert, W. Extending the Host Range of Listeria monocytogenes by Rational Protein Design. Cell 2007, 129, 891–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orazio, S.E.F. Animal models for oral transmission of Listeria monocytogenes. Front. Cell. Infect. Microbial. 2014, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Schlüter, D.; Vuckovic, D.; Wraber, B.; Doric, M.; Deckert, M. Murine model of pregnancy-associated Listeria monocytogenes infection. FEMS Immunol. Med. Microbiol. 2003, 35, 177–182. [Google Scholar] [CrossRef]
- Gessain, G.; Tsai, Y.; Travier, L.; Bonazzi, M.; Grayo, S.; Cossart, P.; Charlier, C.; Disson, O.; Lecuit, M. PI3-kinase activation is critical for host barrier permissiveness to Listeria monocytogenes. J. Exp. Med. 2015, 212, 165–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, K.P.; Faith, N.G.; Steinberg, H.; Czuprynski, C.J. Pregnancy reduces the genetic resistance of C57BL/6 mice to Listeria monocytogenes infection by intragastric inoculation. Microb. Pathog. 2011, 50, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Barber, E.M.; Fazzari, M.; Pollard, J.W. Th1 Cytokines Are Essential for Placental Immunity to Listeria monocytogenes. Infect. Immun. 2005, 73, 6322–6331. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W.; Guleria, I. The trophoblast is a component of the innate immune system during pregnancy. Nat. Med. 2000, 6, 589–593. [Google Scholar] [CrossRef]
- Rowe, J.; Ertelt, J.; Aguilera, M.; Farrar, M.; Way, S. Foxp3+ Regulatory T Cell Expansion Required for Sustaining Pregnancy Compromises Host Defense against Prenatal Bacterial Pathogens. Cell Host Microbe 2011, 10, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Nelson, D.M.; Smith, S.D.; Khun, H.; Huerre, M.; Vacher-Lavenu, M.; Gordon, J.I.; Cossart, P.; Jacob, F. Targeting and Crossing of the Human Maternofetal Barrier by Listeria monocytogenes: Role of Internalin Interaction with Trophoblast E-Cadherin. Proc. Natl. Acad. Sci. USA 2004, 101, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Khelef, N.; Lecuit, M.; Bierne, H.; Cossart, P. Species specificity of the Listeria monocytogenes InlB protein. Cell. Microbiol. 2006, 8, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.V.; Floyd, A.D. Placental development in the Mongolian gerbil (Meriones unguiculatus). I. Early development to the time of chorio-allantoic contact. Am. J. Anat. 1972, 134, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Dussurget, O.; Cossart, P.; Grayo, S.; Disson, O.; Nikitas, G. Modeling human listeriosis in natural and genetically engineered animals. Nat. Protoc. 2009, 4, 799–810. [Google Scholar] [CrossRef]
- Ireton, K.; Cossart, P. Host-Pathogen Interactions During Entry And Actin-based Movement of Listeria monocytogenes. Ann. Rev. Genet. 1997, 31, 113–138. [Google Scholar] [CrossRef] [PubMed]
- Zeldovich, V.B.; Clausen, C.H.; Bradford, E.; Fletcher, D.A.; Maltepe, E.; Robbins, J.R.; Bakardjiev, A.I. Placental syncytium forms a biophysical barrier against pathogen invasion. PLoS Pathog. 2013, 9, e1003821. [Google Scholar] [CrossRef] [PubMed]
- Zeldovich, V.B.; Bakardjiev, A.I. Host Defense and Tolerance: Unique Challenges in the Placenta. PLoS Pathog. 2012, 8, e1002804. [Google Scholar] [CrossRef] [PubMed]
- Ireton, K. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell. Microbiol. 2007, 9, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Bierne, H.; Cossart, P. InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J. Cell Sci. 2002, 115, 3357–3367. [Google Scholar] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Faralla, C.; Rizzuto, G.A.; Lowe, D.E.; Kim, B.; Cooke, C.; Shiow, L.R.; Bakardjiev, A.I. InlP, a New Virulence Factor with Strong Placental Tropism. Infect. Immun. 2016, 84, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Zeldovich, V.B.; Robbins, J.R.; Kapidzic, M.; Lauer, P.; Bakardjiev, A.I. Invasive Extravillous Trophoblasts Restrict Intracellular Growth and Spread of Listeria monocytogenes. PLoS Pathog. 2011, 7, e1002005. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Portnoy, D.A. Listeriolysin O: A phagosome-specific lysin. Microbes Infect. 2007, 9, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Le Monnier, A.; Autret, N.; Join-Lambert, O.F.; Jaubert, F.; Charbit, A.; Berche, P.; Kayal, S. ActA Is Required for Crossing of the Fetoplacental Barrier by Listeria monocytogenes. Infect. Immun. 2007, 75, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Bakardjiev, A.I.; Stacy, B.A.; Portnoy, D.A. Growth of Listeria monocytogenes in the Guinea Pig Placenta and Role of Cell-to-Cell Spread in Fetal Infection. J. Infect. Dis. 2005, 191, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, A. Immunology of the Maternal-Fetal Interface. Ann. Rev. Immunol. 2013, 31, 387–411. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Cardenas, I. The Immune System in Pregnancy: A Unique Complexity. Am. J. Reprod. Immunol. 2010, 63, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Weetman, A.P. The immunology of pregnancy. Thyroid 1999, 9, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, C.A.; Redman, C.W.; Stirrat, G.M. HLA A, B, C antigens are expressed on nonvillous trophoblast of the early human placenta. J. Immunol. 1981, 127, 2614–2616. [Google Scholar] [PubMed]
- Rowe, J.H.; Ertelt, J.M.; Xin, L.; Way, S.S. Listeria monocytogenes Cytoplasmic Entry Induces Fetal Wastage by Disrupting Maternal Foxp3+ Regulatory T Cell-Sustained Fetal Tolerance. PLoS Pathog. 2012, 8, e1002873. [Google Scholar] [CrossRef] [PubMed]
- Raghupathy, R. Th1-type immunity is incompatible with successful pregnancy. Immunol. Today 1997, 18, 478–482. [Google Scholar] [CrossRef]
- Wegmann, T.G.; Lin, H.; Guilbert, L.; Mosmann, T.R. Bidirectional cytokine interactions in the maternal-fetal relationship: Is successful pregnancy a TH2 phenomenon? Immunol. Today 1993, 14, 353–356. [Google Scholar] [CrossRef]
- Chaouat, G.; Zourbas, S.; Ostojic, S.; Lappree-Delage, G.; Dubanchet, S.; Ledee, N.; Martal, J. A brief review of recent data on some cytokine expressions at the materno-foetal interface which might challenge the classical Th1/Th2 dichotomy. J. Reprod. Immunol. 2002, 53, 241–256. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Shen, H. Innate and adaptive immune responses to Listeria monocytogenes: A short overview. Microbes Infect. 2007, 9, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 2004, 4, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Dussurget, O.; Bierne, H.; Cossart, P. The bacterial pathogen Listeria monocytogenes and the interferon family: Type I, type II and type III interferons. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E. Immunity to Intracellular Bacteria. Ann. Rev. Immunol. 1993, 11, 129–163. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.R.; Orlofsky, A.; Pollard, J.W.; Arceci, R.; Ladner, M.B.; Bartocci, A. Apparent role of the macrophage growth factor, CSF-1, in placental development. Nature 1987, 330, 484–486. [Google Scholar] [CrossRef]
- Arceci, R.J.; Shanahan, F.; Stanley, E.R.; Pollard, J.W. Temporal Expression and Location of Colony-Stimulating Factor 1 (CSF-1) and Its Receptor in the Female Reproductive Tract are Consistent with CSF-1-Regulated Placental Development. Proc. Natl. Acad. Sci. USA 1989, 86, 8818–8822. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhu, L.; Pollard, J.W. Colony-Stimulating Factor-1-Dependent Macrophage Functions Regulate the Maternal Decidua Immune Responses against Listeria monocytogenes Infections during Early Gestation in Mice. Infect. Immun. 2009, 77, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, E.M.; Pollard, J.W. The Uterine NK Cell Population Requires IL-15 but These Cells Are Not Required for Pregnancy nor the Resolution of a Listeria monocytogenes Infection. J. Immunol. 2003, 171, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Rizzutoa, G.; Taglianic, E.; Manandhard, P.; Erlebacher, A.; Bakardjiev, A.I. Limited Colonization Undermined by Inadequate Early Immune Responses Defines the Dynamics of Decidual Listeriosis. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.F.; Knubel, C.P.; Sanchez, M.C.; Cervi, L.; Motran, C.C. Pregnancy-specific glycoprotein 1a activates dendritic cells to provide signals for Th17-, Th2-, and Treg-cell polarization. Eur. J. Immunol. 2012, 42, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Nancy, P.; Tagliani, E.; Tay, C.; Asp, P.; Levy, D.E.; Erlebacher, A. Chemokine Gene Silencing in Decidual Stromal Cells Limits T Cell Access to the Maternal-Fetal Interface. Science 2012, 336, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Ertelt, J.M.; Rowe, J.H.; Mysz, M.A.; Singh, C.; Roychowdhury, M.; Aguilera, M.N.; Way, S.S. Foxp3+ Regulatory T Cells Impede the Priming of Protective CD8+ T Cells. J. Immunol. 2011, 187, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Ertelt, J.M.; Jiang, T.T.; Kinder, J.M.; Xin, L.; Owens, K.J.; Jones, H.N. CXCR3 blockade protects against Lm infection-induced fetal wastage. J. Clin. Investig. 2015, 125, 1713–1725. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamond, N.M.; Freitag, N.E. Vertical Transmission of Listeria monocytogenes: Probing the Balance between Protection from Pathogens and Fetal Tolerance. Pathogens 2018, 7, 52. https://doi.org/10.3390/pathogens7020052
Lamond NM, Freitag NE. Vertical Transmission of Listeria monocytogenes: Probing the Balance between Protection from Pathogens and Fetal Tolerance. Pathogens. 2018; 7(2):52. https://doi.org/10.3390/pathogens7020052
Chicago/Turabian StyleLamond, Nicole M., and Nancy E. Freitag. 2018. "Vertical Transmission of Listeria monocytogenes: Probing the Balance between Protection from Pathogens and Fetal Tolerance" Pathogens 7, no. 2: 52. https://doi.org/10.3390/pathogens7020052
APA StyleLamond, N. M., & Freitag, N. E. (2018). Vertical Transmission of Listeria monocytogenes: Probing the Balance between Protection from Pathogens and Fetal Tolerance. Pathogens, 7(2), 52. https://doi.org/10.3390/pathogens7020052