VCGIDB: A Database and Web Resource for the Genomic Islands from Vibrio cholerae



Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
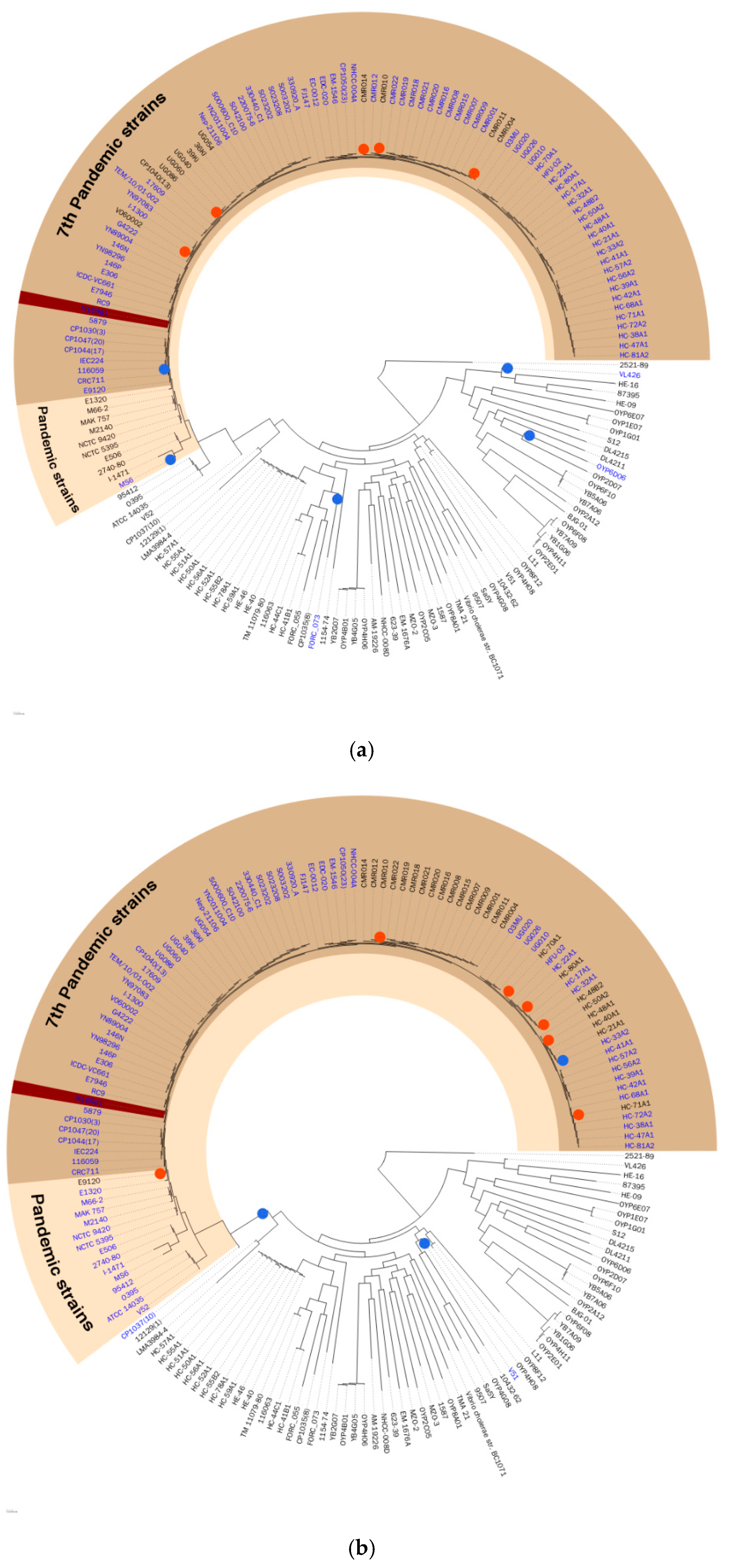
4.1. Genomes and Phylogeny of V. Cholerae
4.2. Detecting Candidate GIs with GI-Scanner
4.3. Identification and Annotation of Genomic Islands
4.4. Annotation
4.5. Testing the Expansion Capability of the Database
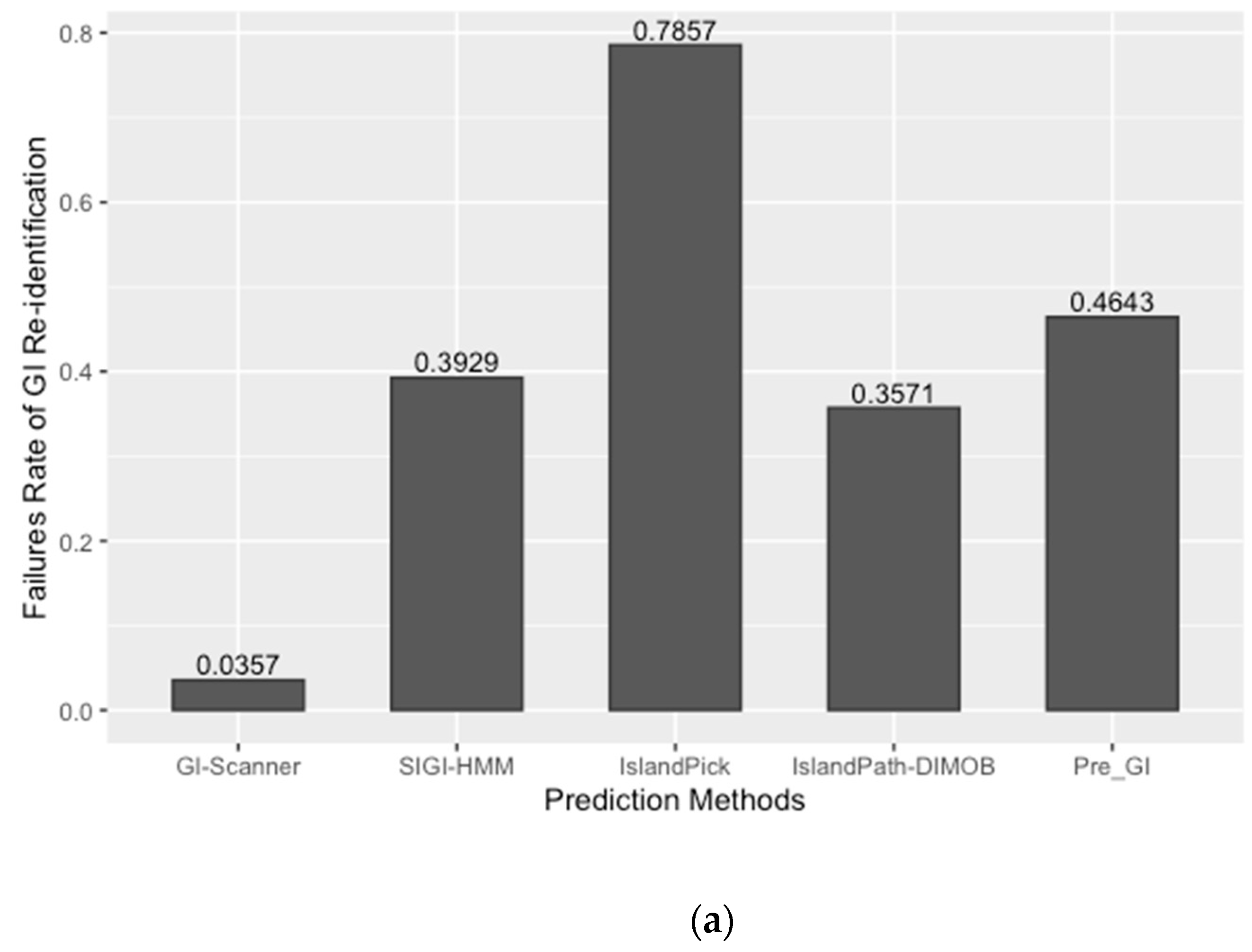
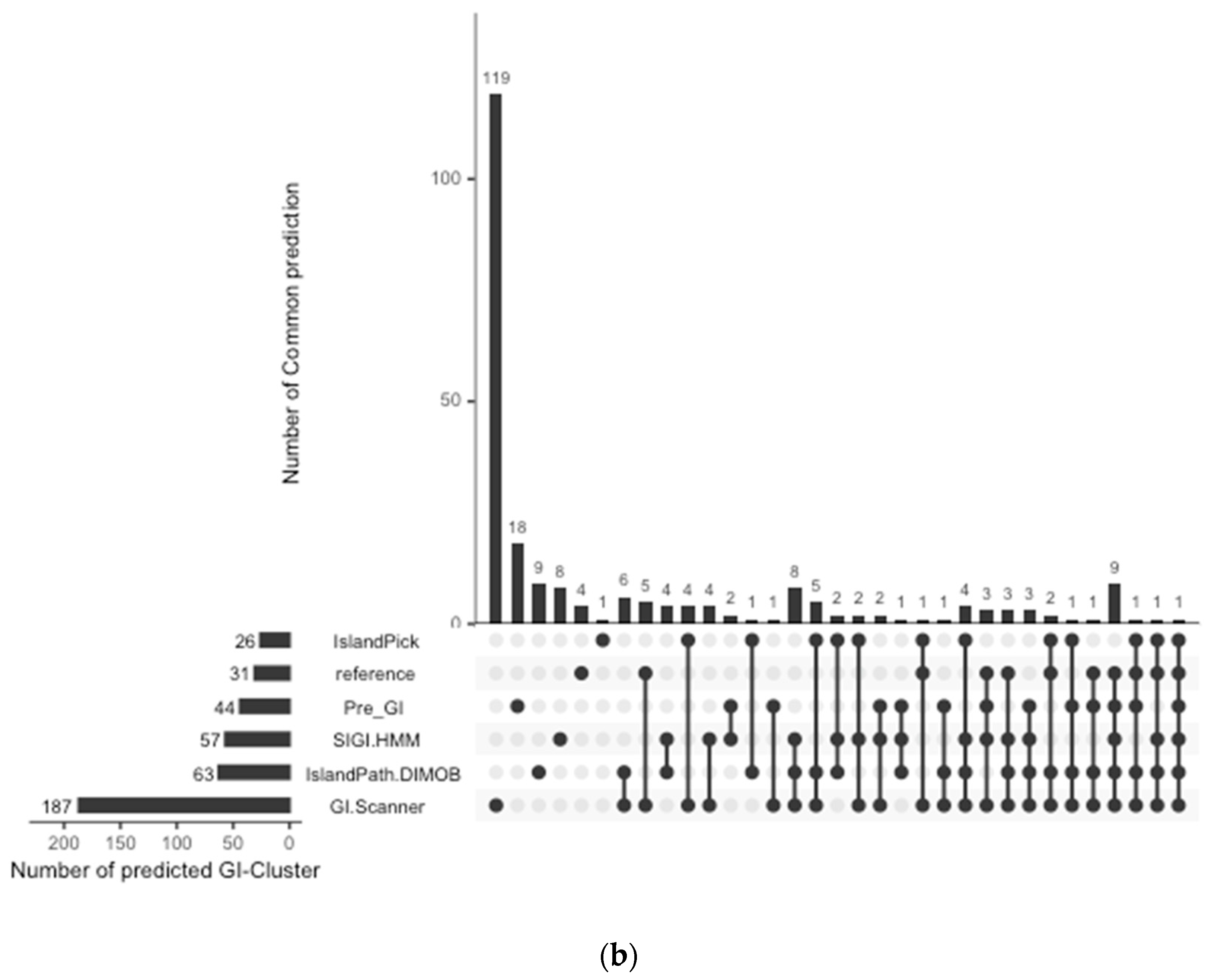
4.6. Database Comparison and Validation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sack, D.A.; Sack, R.B.; Nair, G.B.; Siddique, A.K. Cholera. Lancet 2004, 363, 223–233. [Google Scholar] [CrossRef]
- Chapman, C.; Henry, M.; Bishop-Lilly, K.A.; Awosika, J.; Briska, A.; Ptashkin, R.N.; Wagner, T.; Rajanna, C.; Tsang, H.; Johnson, S.L.; et al. Scanning the Landscape of Genome Architecture of Non-O1 and Non-O139 Vibrio cholerae by Whole Genome Mapping Reveals ExtensivePopulation Genetic Diversity. PLoS ONE 2015, 10, e0120311. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, G.H.; Mahalanabis, D. New strains of Vibrio cholerae O139 in India and Bangladesh: Lessonsfrom the recent epidemics. J. Diarrhoeal Dis. Res. 1993, 11, 63–66. [Google Scholar] [PubMed]
- Cho, Y.J.; Yi, H.; Lee, J.H.; Kim, D.W.; Chun, J. Genomic evolution of Vibrio cholerae. Curr. Opin. Microbiol. 2010, 13, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.W.L.; Ung, K.; Aeschliman, D.; Bryan, J.; Finlay, B.B.; Brinkman, F.S.L. Evidence of a Large Novel Gene Pool Associated with Prokaryotic Genomic Islands. PLoS Genet. 2005, 1, e62. [Google Scholar] [CrossRef]
- Chun, J.; Grim, C.J.; Hasan, N.A.; Lee, J.H.; Choi, S.Y.; Haley, B.J.; Taviani, E.; Jeon, Y.S.; Kim, D.W.; Lee, J.H.; et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2009, 106, 15442–15447. [Google Scholar] [CrossRef]
- Kaas, R.S.; Ngandjio, A.; Nzouankeu, A.; Siriphap, A.; Fonkoua, M.C.; Aarestrup, F.M.; Hendriksen, R.S. The Lake Chad Basin, an Isolated and Persistent Reservoir of Vibrio cholerae O1: A Genomic Insight into the Outbreak in Cameroon, 2010. PLoS ONE 2016, 11, e0155691. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands forlarger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Yoon, S.H.; Park, Y.K.; Kim, J.F. PAIDB v2.0: Exploration and analysis of pathogenicity and resistanceislands. Nucleic Acids Res. 2014, 43, D624–D630. [Google Scholar] [CrossRef]
- Pierneef, R.; Cronje, L.; Bezuidt, O.; Reva, O.N. Pre_GI: A global map of ontological links between horizontally transferred genomic islands in bacterial and archaeal genomes. Database 2015, 2015, bav058. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Hsiao, W.W.L.; Brinkman, F.S.L. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 2008, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S.L. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef]
- Lu, B.; Leong, H.W. Computational methods for predicting genomic islands in microbial genomes. Comput. Struct. Biotechnol. J. 2016, 14, 200–206. [Google Scholar] [CrossRef]
- Dziejman, M.; Balon, E.; Boyd, D.; Fraser, C.M.; Heidelberg, J.F.; Mekalanos, J.J. Comparative genomic analysis of Vibrio cholerae: Genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. USA 2002, 99, 1556–1561. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evolut. Microbiol. 2017, 67, 1613–1617. [Google Scholar]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. bioRxiv 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evolut. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Kannan, L.; Wheeler, W.C. Maximum Parsimony on Phylogenetic networks. Algorithms Mol. Biol. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Zeevi, D.; Korem, T.; Godneva, A.; Bar, N.; Kurilshikov, A.; Lotan-Pompan, M.; Weinberger, A.; Fu, J.; Wijmenga, C.; Zhernakova, A.; et al. Structural variation in the gut microbiome associates with host health. Nature 2019, 568, 43. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Assembly Level | Accumulated Number of Strains | Accumulated Number of Predicted GIs |
|---|---|---|
| Complete or Chromosome | 58 | 198 |
| Scaffold | 277 | 312 |
| Contig | 798 | 435 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, Y.; Chalita, M.; Ha, S.-m.; Baek, I.; Chun, J. VCGIDB: A Database and Web Resource for the Genomic Islands from Vibrio cholerae. Pathogens 2019, 8, 261. https://doi.org/10.3390/pathogens8040261
Hur Y, Chalita M, Ha S-m, Baek I, Chun J. VCGIDB: A Database and Web Resource for the Genomic Islands from Vibrio cholerae. Pathogens. 2019; 8(4):261. https://doi.org/10.3390/pathogens8040261
Chicago/Turabian StyleHur, YoungJae, Mauricio Chalita, Sung-min Ha, Inwoo Baek, and Jongsik Chun. 2019. "VCGIDB: A Database and Web Resource for the Genomic Islands from Vibrio cholerae" Pathogens 8, no. 4: 261. https://doi.org/10.3390/pathogens8040261
APA StyleHur, Y., Chalita, M., Ha, S. -m., Baek, I., & Chun, J. (2019). VCGIDB: A Database and Web Resource for the Genomic Islands from Vibrio cholerae. Pathogens, 8(4), 261. https://doi.org/10.3390/pathogens8040261