Altered Salivary Microbiome in the Early Stage of HIV Infections among Young Chinese Men Who Have Sex with Men (MSM)

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Study Cohort
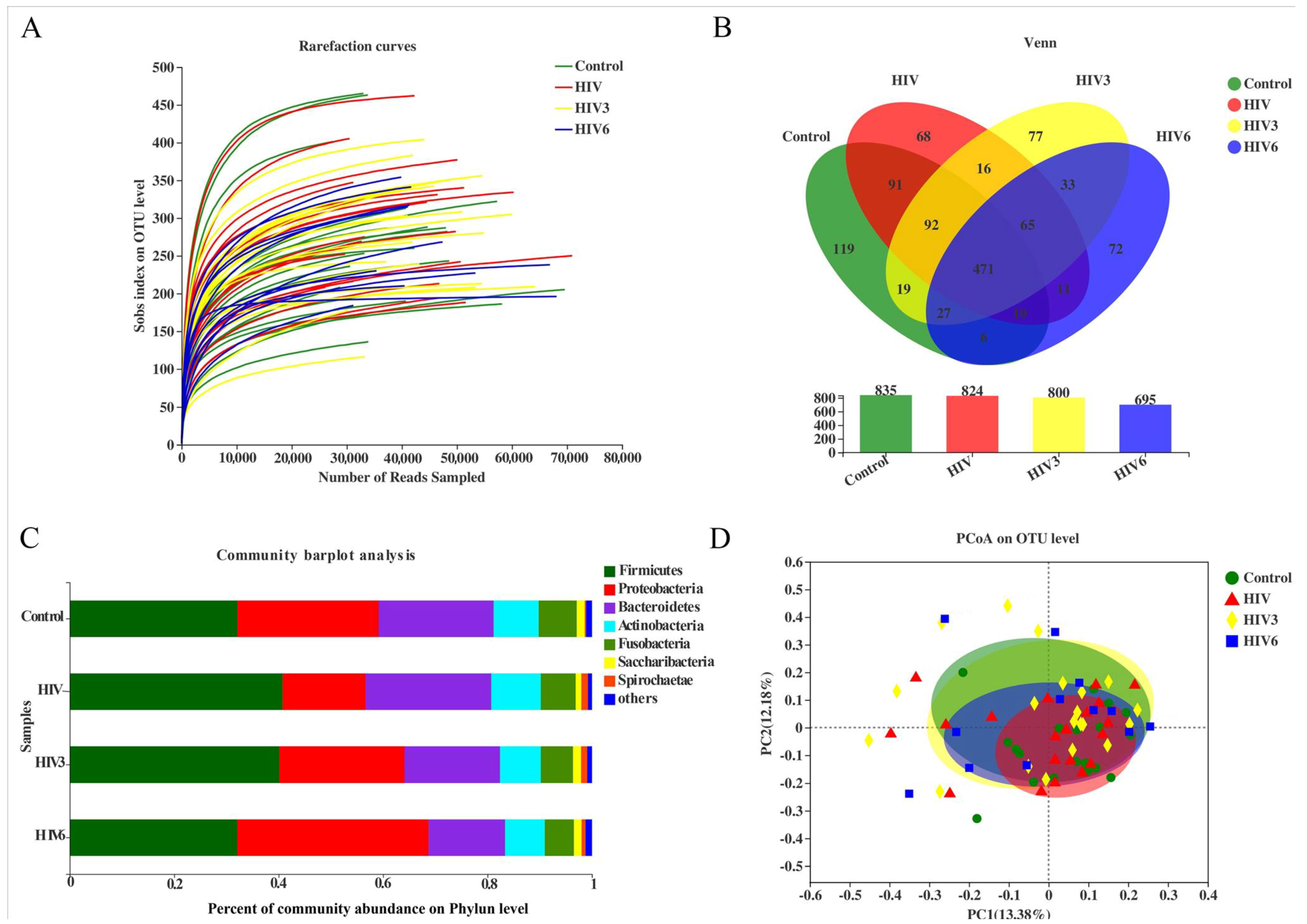
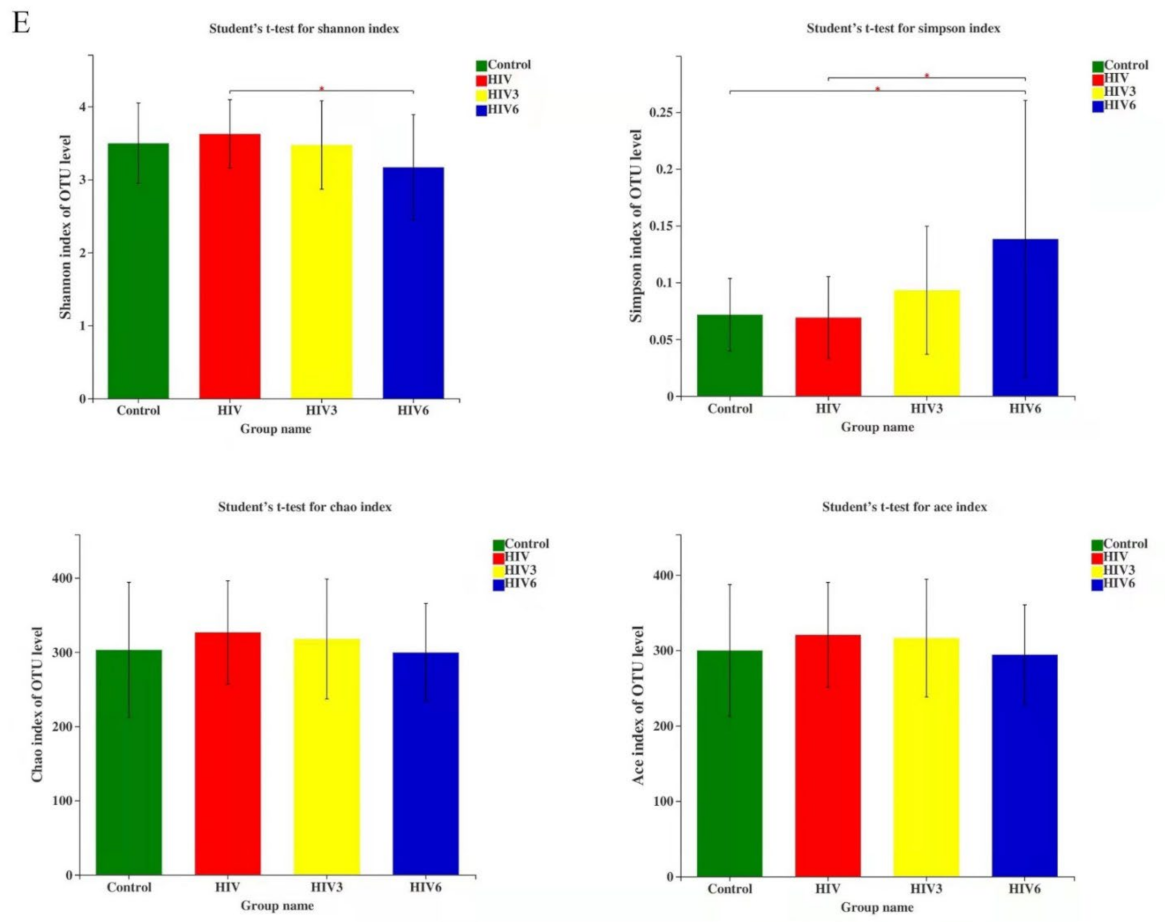
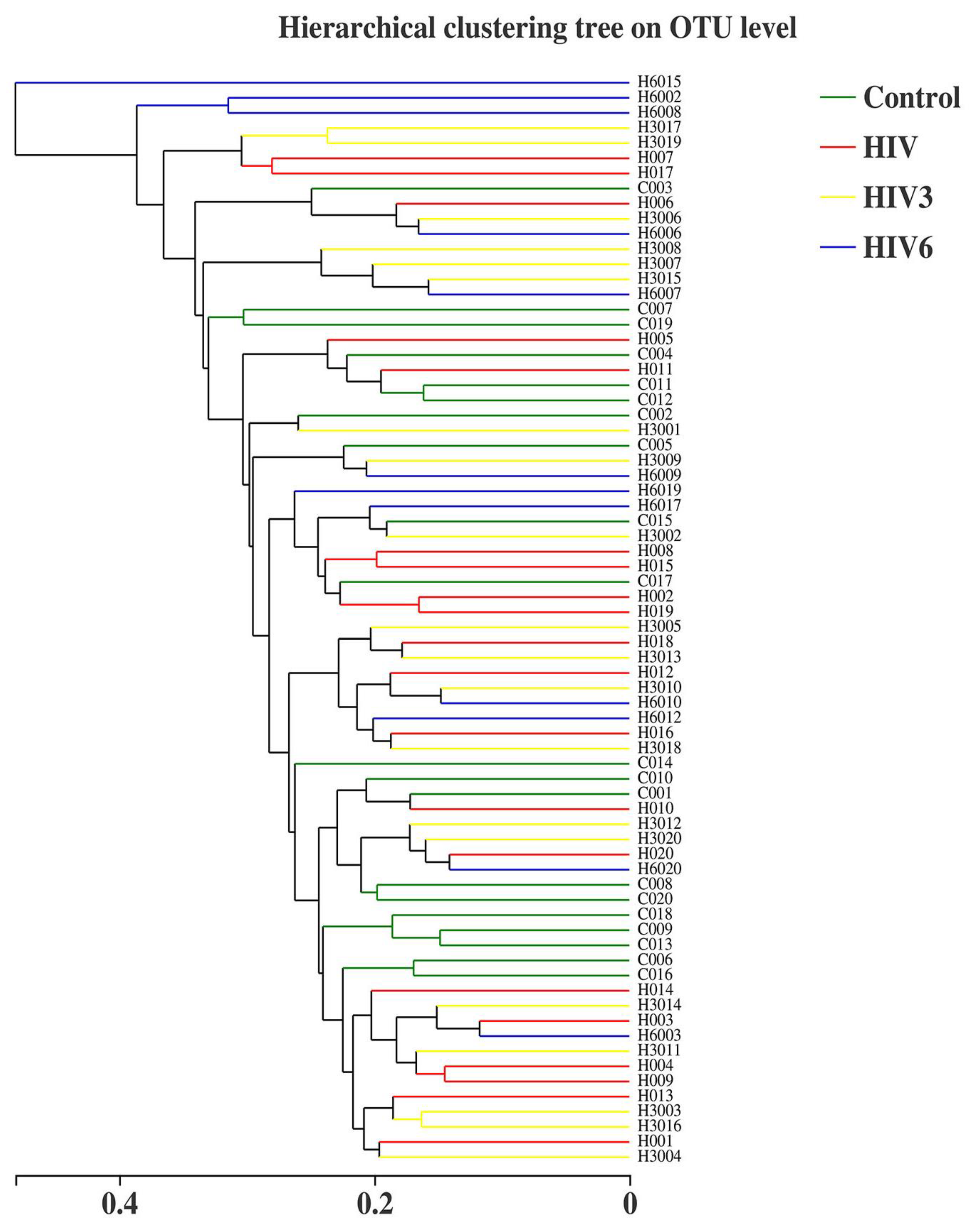
2.2. Salivary Microbiome Diversity between Healthy Controls and HIV Infections over the Course of ART
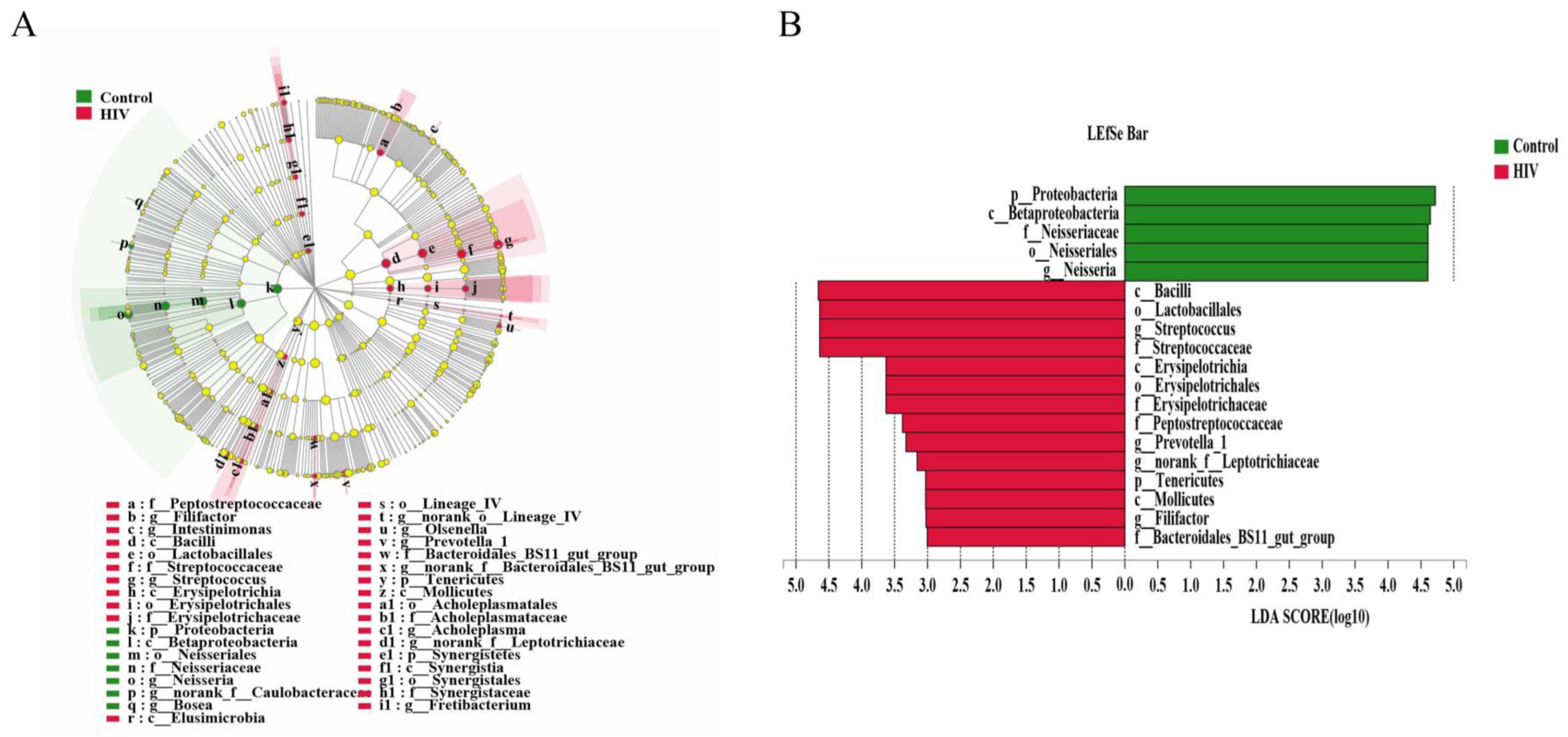
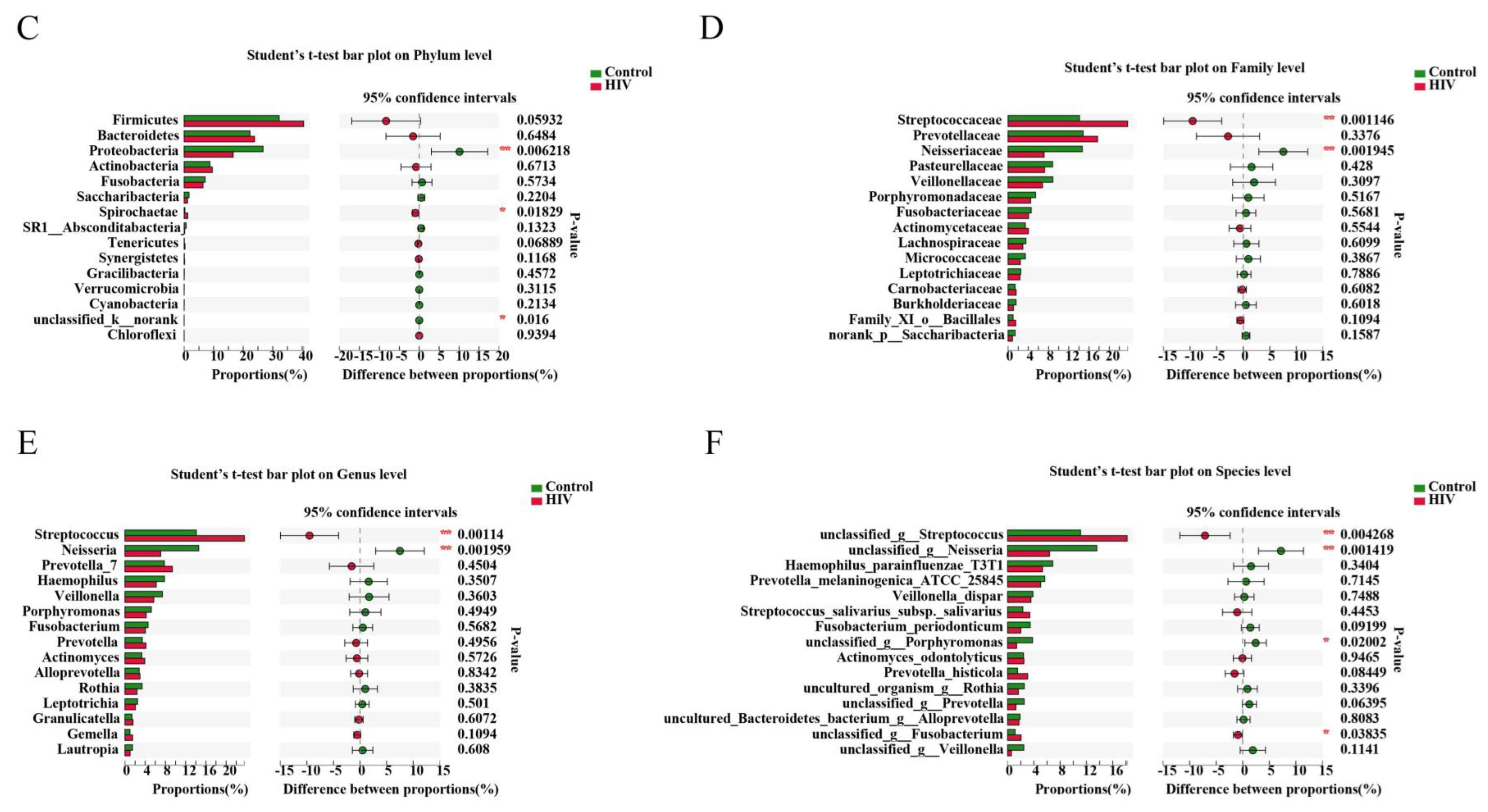
2.3. Different Salivary Microbiome between HIV Infections before ART and HIV-Uninfected Group
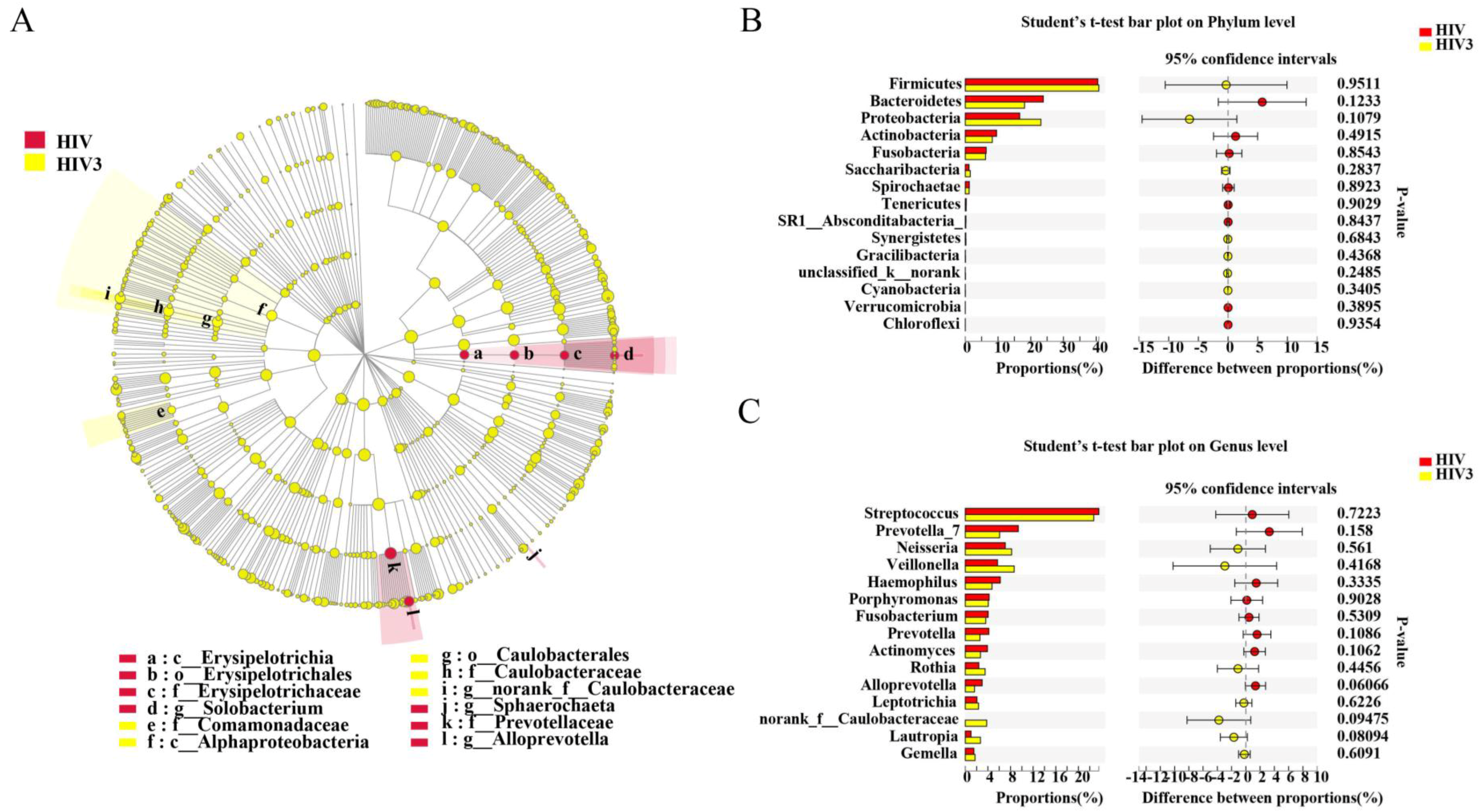
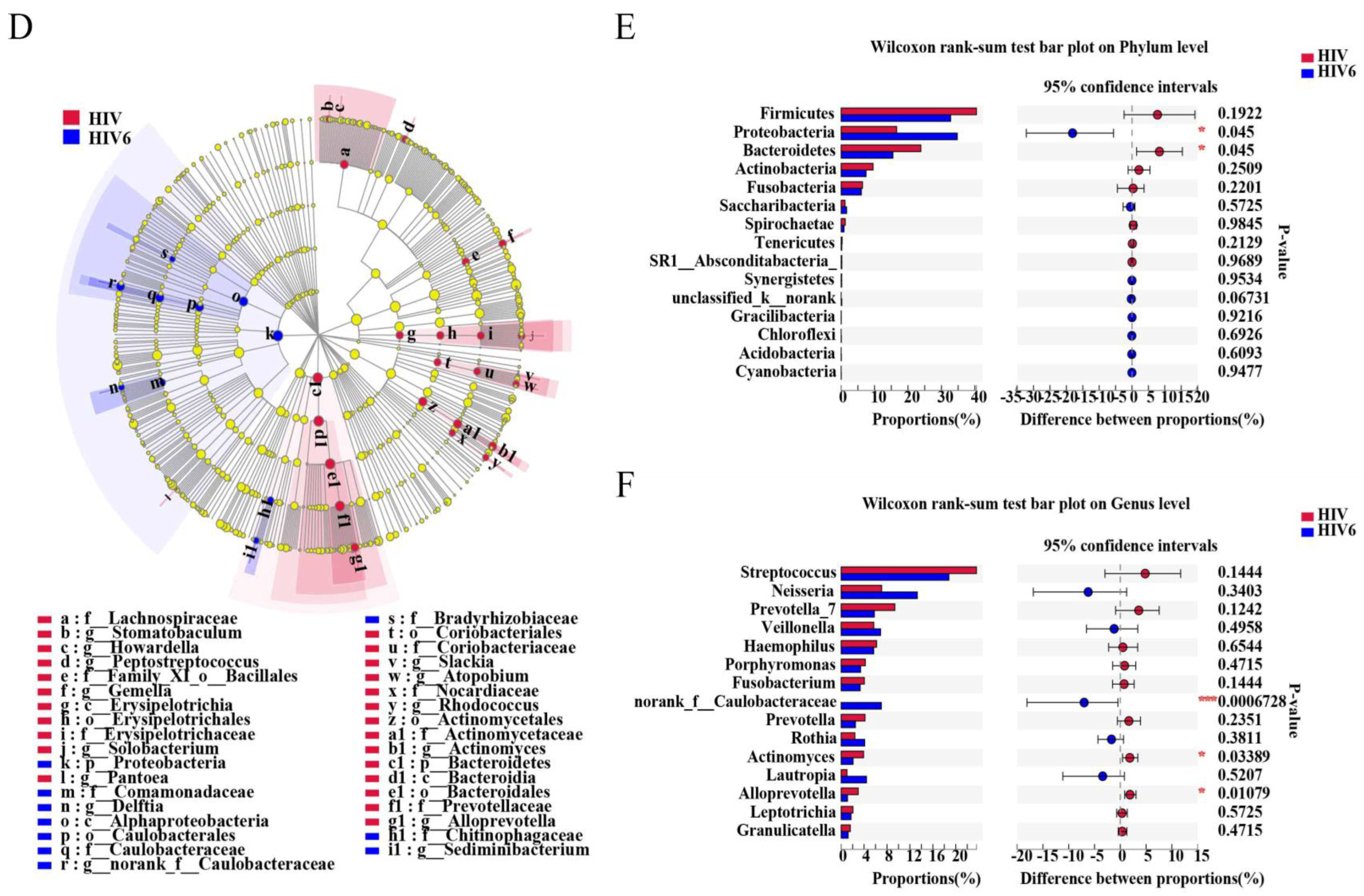
2.4. Impact of Three and Six Months of ART on the Salivary Microbiome in HIV Infections
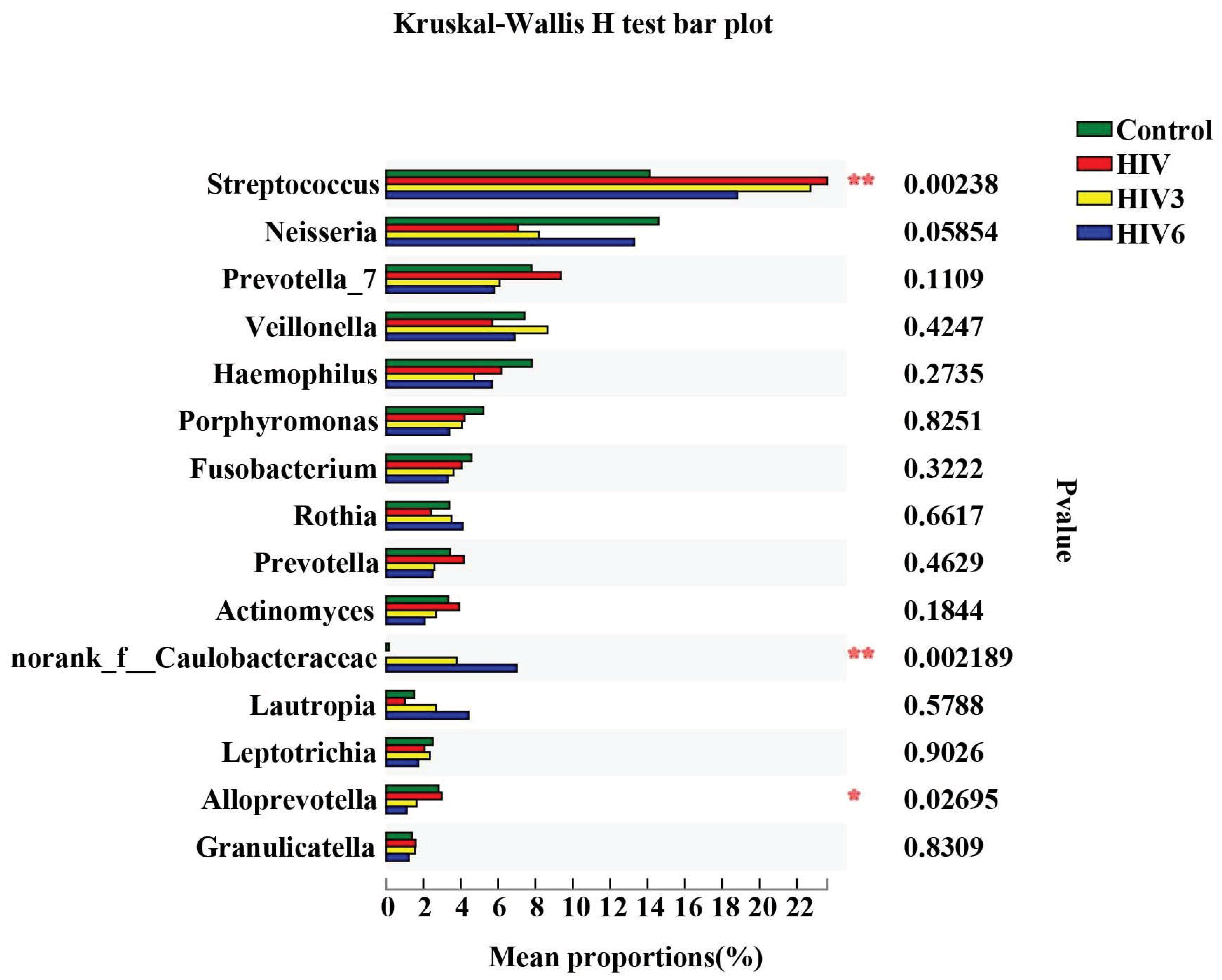
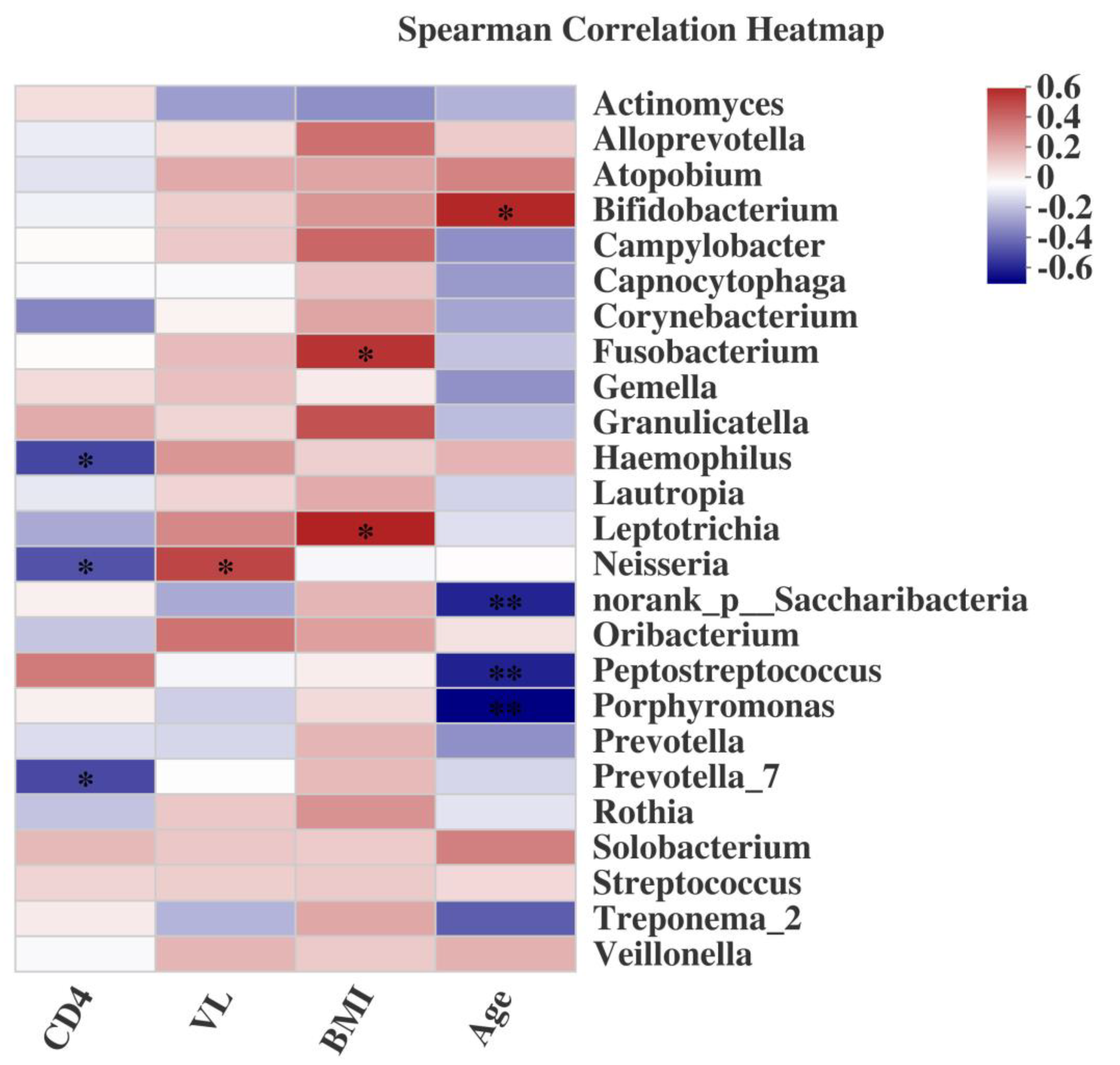
2.5. Associations of the Salivary Microbiome with Demographic Characteristics, CD4+ T cell Count, and VL
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Sample Collection and Initial Processing
4.3. Genomic DNA Isolation and PCR Amplification
4.4. Illumina MiSeq Sequencing
4.5. Data Processing and Sequence Analysis
4.6. Statistical Analysis
4.7. Ethics Statement
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Available online: http://www.who.int/en/news-room/fact-sheets/detail/hiv-aids (accessed on 6 July 2020).
- McLaughlin, K. HIV infections are spiking among young gay Chinese. Science 2017, 355, 1359. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, J.S.; Barr, C.E.; Sciubba, J.J.; Winkler, J.R. Oral manifestations of HIV infection. Definitions, diagnostic criteria, and principles of therapy. The U.S.A. Oral AIDS Collaborative Group. Oral Surg. Oral Med. Oral Pathol. 1992, 73, 142–144. [Google Scholar] [CrossRef]
- Reznik, D.A. Oral manifestations of HIV disease. Top. HIV Med. 2005, 13, 143–148. [Google Scholar]
- Greenspan, J.S. Sentinels and signposts: The epidemiology and significance of the oral manifestations of HIV disease. Oral Dis. 1997, 3, S13–S17. [Google Scholar] [CrossRef]
- Moyes, D.L.; Saxena, D.; John, M.D.; Malamud, D. The gut and oral microbiome in HIV disease: A workshop report. Oral Dis. 2016, 22, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Nittayananta, W.; Tao, R.; Jiang, L.; Peng, Y.; Huang, Y. Oral innate immunity in HIV infection in HAART era. J. Oral Pathol. Med. 2016, 45, 3–8. [Google Scholar] [CrossRef] [Green Version]
- WHO. What’s New in Treatment Monitoring: Viral Load and CD4 Testing. 2017. Available online: http://www.who.int/hiv/pub/arv/treatment-monitoring-info-2017/en/2017 (accessed on 6 July 2017).
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Wade, W.G. The oral microbiome in health and disease. Pharm. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Luo, Y.; Yuan, L.; Nelson, K.E.; Wang, Y.; Xiang, C.; Li, L. Pyrosequencing analysis of the human microbiota of healthy Chinese undergraduates. BMC Genom. 2013, 14, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistler, J.O.; Arirachakaran, P.; Poovorawan, Y.; Dahlen, G.; Wade, W.G. The oral microbiome in human immunodeficiency virus (HIV)-positive individuals. J. Med. Microbiol. 2015, 64, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, S.K.; Uppal, K.; Li, S.; Jones, D.P.; Huang, L.; Tipton, L.; Fitch, A.; Greenblatt, R.M.; Kingsley, L.; Guidot, D.M.; et al. Correlation of the lung microbiota with metabolic profiles in bronchoalveolar lavage fluid in HIV infection. Microbiome 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Li, M.; Campbell, T.B.; Flores, S.C.; Linderman, D.; Gebert, M.J.; Knight, R.; Fontenot, A.P.; Palmer, B.E. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 2013, 14, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J. Infectious Disease. Vaginal microbiome affects HIV risk. Science 2016, 353, 331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Cheng, X.Q.; Li, J.Y.; Zhang, P.; Yi, P.; Xu, X.; Zhou, X.D. Saliva in the diagnosis of diseases. Int. J. Oral Sci. 2016, 8, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Gesime, J.; Luciano, R. Saliva as a Diagnostic Tool for Oral and Systemic Diseases. Literature Review. Acta Bioclinica 2018, 8, 188–211. [Google Scholar]
- Liu, J.Y.; Duan, Y.X. Saliva: A potential media for disease diagnostics and monitoring. Oral Oncol. 2012, 48, 569–577. [Google Scholar] [CrossRef]
- Kutsch, V.K.; Young, D.A. New directions in the etiology of dental caries disease. J. Calif. Dent. Assoc. 2011, 39, 716–721. [Google Scholar]
- Prakasam, S.; Srinivasan, M. Evaluation of salivary biomarker profiles following non-surgical management of chronic periodontitis. Oral Dis. 2014, 20, 171–177. [Google Scholar] [CrossRef]
- Zhong, L.P.; Zhang, C.P.; Zheng, J.W.; Li, J.; Chen, W.T.; Zhang, Z.Y. Increased Cyfra 21-1 concentration in saliva from primary oral squamous cell carcinoma patients. Arch. Oral Biol. 2007, 52, 1079–1087. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Li, Y.; Saxena, D.; Chen, Z.; Liu, G.; Abrams, W.R.; Phelan, J.A.; Norman, R.G.; Fisch, G.S.; Corby, P.M.; Dewhirst, F.; et al. HIV infection and microbial diversity in saliva. J. Clin. Microbiol. 2014, 52, 1400–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.M.; Schloss, P.D.; Venkataraman, A.; Twigg, H., 3rd; Jablonski, K.A.; Bushman, F.D.; Campbell, T.B.; Charlson, E.S.; Collman, R.G.; Crothers, K.; et al. Multicenter Comparison of Lung and Oral Microbiomes of HIV-infected and HIV-uninfected Individuals. Am. J. Respir. Crit. Care Med. 2015, 192, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Salas, J.T.; Chang, T.L. Microbiome in human immunodeficiency virus infection. Clin. Lab. Med. 2014, 34, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, D.; Li, Y.; Yang, L.; Pei, Z.; Poles, M.; Abrams, W.R.; Malamud, D. Human microbiome and HIV/AIDS. Curr. HIV/AIDS Rep. 2012, 9, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.K.; Chandra, J.; Retuerto, M.; Sikaroodi, M.; Brown, R.E.; Jurevic, R.; Salata, R.A.; Lederman, M.M.; Gillevet, P.M.; Ghannoum, M.A. Oral mycobiome analysis of HIV-infected patients: Identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 2014, 10, e1003996. [Google Scholar] [CrossRef] [PubMed]
- Gaester, K.; Fonseca, L.A.M.; Luiz, O.; Assone, T.; Fontes, A.S.; Costa, F.; Duarte, A.J.S.; Casseb, J. Human papillomavirus infection in oral fluids of HIV-1-positive men: Prevalence and risk factors. Sci. Rep. 2014, 4, 6592. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, W.; Operario, D.; Zhao, Y.; Shi, A.; Zhang, Z.; Gao, P.; Perez, A.; Wang, J.; Zaller, N.; et al. Effects of a Mobile Health Intervention to Promote HIV Self-testing with MSM in China: A Randomized Controlled Trial. AIDS Behav. 2019, 23, 3129–3139. [Google Scholar] [CrossRef] [Green Version]
- Dang, A.T.; Cotton, S.; Sankaran-Walters, S.; Li, C.S.; Lee, C.Y.; Dandekar, S.; Paster, B.J.; George, M.D. Evidence of an increased pathogenic footprint in the lingual microbiome of untreated HIV infected patients. BMC Microbiol. 2012, 12, 153. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Cardoso, S.; Klatt, N.R.; Reyes-Teran, G. Impact of antiretroviral drugs on the microbiome: Unknown answers to important questions. Curr. Opin. HIV AIDS 2018, 13, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Proctor, D.M.; Fukuyama, J.A.; Loomer, P.M.; Armitage, G.C.; Lee, S.A.; Davis, N.M.; Ryder, M.I.; Holmes, S.P.; Relman, D.A. A spatial gradient of bacterial diversity in the human oral cavity shaped by salivary flow. Nat. Commun. 2018, 9, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navazesh, M.; Mulligan, R.; Karim, R.; Mack, W.J.; Ram, S.; Seirawan, H.; Greenspan, J.; Greenspan, D.; Phelan, J.; Alves, M.; et al. Effect of HAART on salivary gland function in the Women’s Interagency HIV Study (WIHS). Oral Dis. 2009, 15, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presti, R.M.; Handley, S.A.; Droit, L.; Ghannoum, M.; Jacobson, M.; Shiboski, C.H.; Webster-Cyriaque, J.; Brown, T.; Yin, M.T.; Overton, E.T. Alterations in the oral microbiome in HIV-infected participants after antiretroviral therapy administration are influenced by immune status. Aids 2018, 32, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Landay, A.; Presti, R.M. Microbiome alterations in HIV infection a review. Cell. Microbiol. 2016, 18, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Klatt, N.R.; Cheu, R.; Birse, K.; Zevin, A.S.; Perner, M.; Noel-Romas, L.; Grobler, A.; Westmacott, G.; Xie, I.Y.; Butler, J.; et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 2017, 356, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Fukui, Y.; Aoki, K.; Ishii, Y.; Tateda, K. The palatine tonsil bacteriome, but not the mycobiome, is altered in HIV infection. Bmc Microbiol. 2018, 18, 127. [Google Scholar] [CrossRef]
- Yang, L.Y.; Poles, M.A.; Fisch, G.S.; Ma, Y.F.; Nossa, C.; Phelan, J.A.; Pei, Z.H. HIV-induced immunosuppression is associated with colonization of the proximal gut by environmental bacteria. Aids 2016, 30, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Nowak, R.G.; Bentzen, S.M.; Ravel, J.; Crowell, T.A.; Dauda, W.; Ma, B.; Liu, H.; Blattner, W.A.; Baral, S.D.; Charurat, M.E.; et al. Rectal microbiota among HIV-uninfected, untreated HIV, and treated HIV-infected in Nigeria. Aids 2017, 31, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; He, S.; Jin, J.; Dong, G.; Wu, H. Exploring salivary microbiota in AIDS patients with different periodontal statuses using 454 GS-FLX Titanium pyrosequencing. Front. Cell. Infect. Microbiol. 2015, 5, 55. [Google Scholar] [CrossRef] [Green Version]
- Starr, J.R.; Huang, Y.M.; Lee, K.H.; Murphy, C.M.; Moscicki, A.B.; Shiboski, C.H.; Ryder, M.I.; Yao, T.J.; Faller, L.L.; Van Dyke, R.B.; et al. Oral microbiota in youth with perinatally acquired HIV infection. Microbiome 2018, 6, 100. [Google Scholar] [CrossRef]
- Santacruz, A.; Collado, M.C.; Garcia-Valdes, L.; Segura, M.T.; Martin-Lagos, J.A.; Anjos, T.; Marti-Romero, M.; Lopez, R.M.; Florido, J.; Campoy, C.; et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br. J. Nutr. 2010, 104, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewy, T.; Hong, B.Y.; Weiser, B.; Burger, H.; Tremain, A.; Weinstock, G.; Anastos, K.; George, M.D. Oral Microbiome in HIV-Infected Women: Shifts in the Abundance of Pathogenic and Beneficial Bacteria Are Associated with Aging, HIV Load, CD4 Count, and Antiretroviral Therapy. Aids Res. Hum. Retrov. 2019, 35, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV | HIV6 | Control | |
|---|---|---|---|
| Gender | |||
| Men | 20 | 12 | 20 |
| Women | 0 | 0 | 0 |
| Age (mean ± SD) | 26.30 ± 5.41 | 26.30 ± 5.41 | |
| BMI (mean ± SD) | 21.31 ± 3.19 | 22.31 ± 3.04 | |
| Transmission | |||
| Injection Drug Use | 0 | 0 | 0 |
| Heterosexual | 1 | 0 | 0 |
| Homosexual | 19 | 12 | 0 |
| CD4+ T cell count (Range) (cells/μL) | 219–652 | 323–798 | / |
| CD4 ≥ 500 cells/μL | 3 | 10 | / |
| CD4 < 500 cells/μL | 17 | 2 | / |
| Viral Load (copies/mL) (Range) | 5059–8,749,628 | TND–1930 | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chang, S.; Guo, H.; Ji, Y.; Jiang, H.; Ruan, L.; Du, M. Altered Salivary Microbiome in the Early Stage of HIV Infections among Young Chinese Men Who Have Sex with Men (MSM). Pathogens 2020, 9, 960. https://doi.org/10.3390/pathogens9110960
Li J, Chang S, Guo H, Ji Y, Jiang H, Ruan L, Du M. Altered Salivary Microbiome in the Early Stage of HIV Infections among Young Chinese Men Who Have Sex with Men (MSM). Pathogens. 2020; 9(11):960. https://doi.org/10.3390/pathogens9110960
Chicago/Turabian StyleLi, Jin, Shenghua Chang, Haiying Guo, Yaoting Ji, Han Jiang, Lianguo Ruan, and Minquan Du. 2020. "Altered Salivary Microbiome in the Early Stage of HIV Infections among Young Chinese Men Who Have Sex with Men (MSM)" Pathogens 9, no. 11: 960. https://doi.org/10.3390/pathogens9110960
APA StyleLi, J., Chang, S., Guo, H., Ji, Y., Jiang, H., Ruan, L., & Du, M. (2020). Altered Salivary Microbiome in the Early Stage of HIV Infections among Young Chinese Men Who Have Sex with Men (MSM). Pathogens, 9(11), 960. https://doi.org/10.3390/pathogens9110960