Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins



Abstract
:1. Introduction
2. HPV Infection and Human Carcinogenesis
3. Keratinocytes as Mediators of Innate Immunity Against HPV Infection
4. Impact of E6/E7 on the Innate Immune Response
4.1. cGAS/STING/TBK1
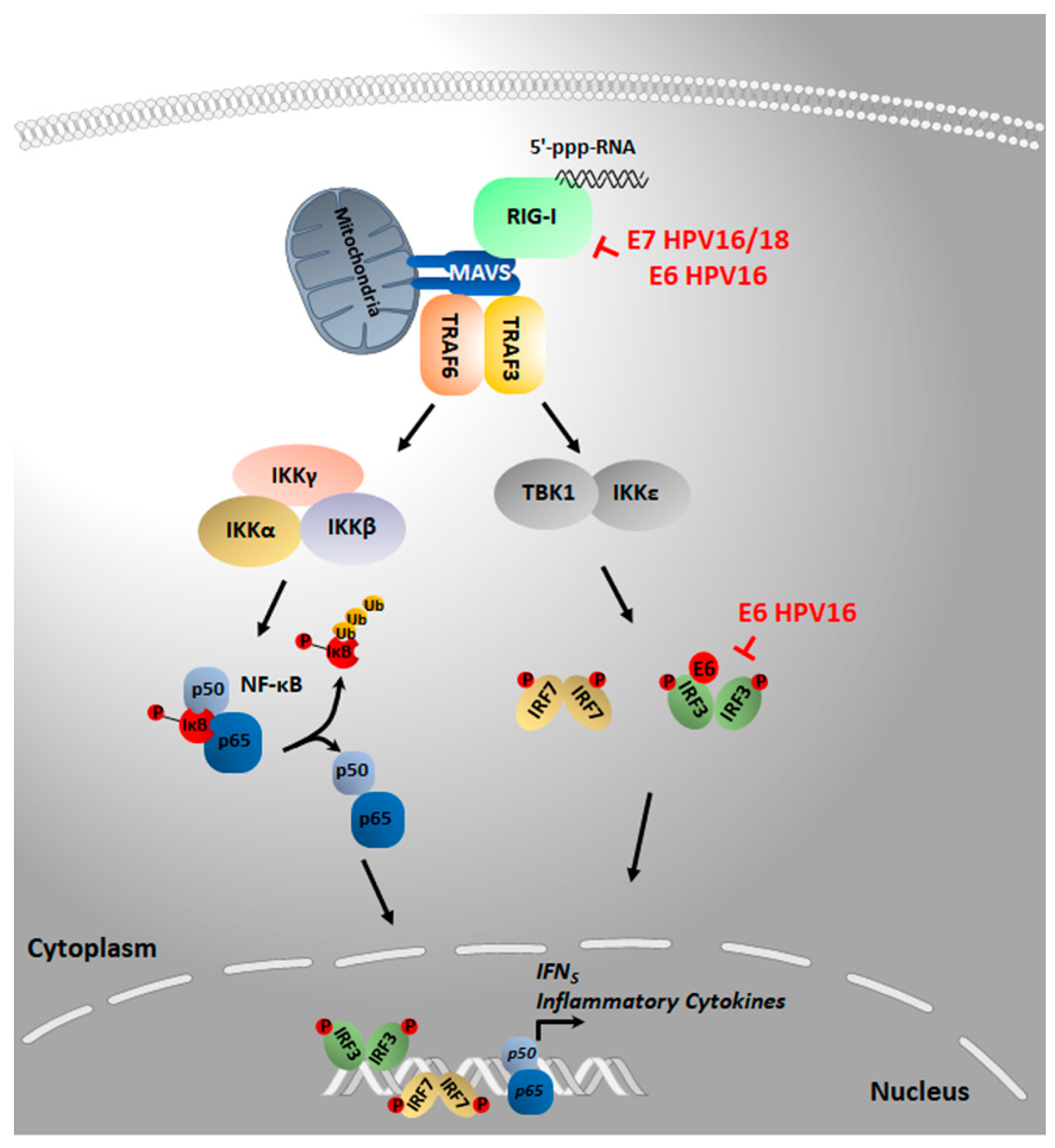
4.2. RIG-I/MAVS/TBK1
4.3. TLRs
4.4. PRR-Related Transcription Factors
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bosch, F.X.; Broker, T.R.; Forman, D.; Moscicki, A.B.; Gillison, M.L.; Doorbar, J.; Stern, P.L.; Stanley, M.; Arbyn, M.; Poljak, M.; et al. Comprehensive control of human papillomavirus infections and related diseases. Vaccine 2013, 31, H1–H31. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K. Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Thomas, M.; Pim, D.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haedicke, J.; Iftner, T. Human papillomaviruses and cancer. Radiother. Oncol. 2013, 108, 397–402. [Google Scholar] [CrossRef]
- Senapati, R.; Senapati, N.N.; Dwibedi, B. Molecular mechanisms of HPV mediated neoplastic progression. Infect. Agent Cancer 2016, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PaVE: The Papillomavirus Knowledge Source. Available online: https://pave.niaid.nih.gov/ (accessed on 10 March 2020).
- Quint, K.D.; Genders, R.E.; de Koning, M.N.; Borgogna, C.; Gariglio, M.; Bouwes Bavinck, J.N.; Doorbar, J.; Feltkamp, M.C. Human Beta-papillomavirus infection and keratinocyte carcinomas. J. Pathol. 2015, 235, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Shewale, J.B.; Gillison, M.L. Dynamic factors affecting HPV-attributable fraction for head and neck cancers. Curr. Opin. Virol. 2019, 39, 33–40. [Google Scholar] [CrossRef]
- Berman, T.A.; Schiller, J.T. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef]
- Edwards, T.G.; Koeller, K.J.; Slomczynska, U.; Fok, K.; Helmus, M.; Bashkin, J.K.; Fisher, C. HPV Episome Levels are Potently Decreased by Pyrrole-Imidazole Polyamides. Antivir. Res. 2011, 91, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA Damage Repair Genes Controlling Human Papillomavirus (HPV) Episome Levels under Conditions of Stability and Extreme Instability. PLoS ONE 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, P.; Aggarwal, A.; Baliyan, V. Complete clearance of cutaneous warts with hydroxychloroquine: Antiviral action? Indian J. Dermatol. 2014, 59, 211. [Google Scholar] [CrossRef]
- Castaneda, C.H.; Scuderi, M.J.; Edwards, T.G.; Harris, G.D., Jr.; Dupureur, C.M.; Koeller, K.J.; Fisher, C.; Bashkin, J.K. Improved Antiviral Activity of a Polyamide Against High-Risk Human Papillomavirus Via N-Terminal Guanidinium Substitution. Med. Chem. Comm. 2016, 7, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Zwerschke, W.; Jansen-Dürr, P. Cell transformation by the E7 oncoprotein of human papillomavirus type 16: Interactions with nuclear and cytoplasmic target proteins. Adv. Cancer Res. 2000, 78, 1–29. [Google Scholar] [CrossRef]
- Tungteakkhun, S.S.; Duerksen-Hughes, P.J. Cellular binding partners of the human papillomavirus E6 protein. Arch. Virol. 2008, 153, 397–408. [Google Scholar] [CrossRef] [Green Version]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [Green Version]
- White, E.A.; Howley, P.M. Proteomic approaches to the study of papillomavirus-host interactions. Virology 2013, 435, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C. Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation. J. Clin. Med. 2015, 4, 204–230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Sparrer, K.M.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Ni, G.; Damania, B. Innate Sensing of DNA Virus Genomes. Annu. Rev. Virol. 2018, 5, 341–362. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479, 146–152. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R. Activation and regulation of DNA-driven immune responses. Microbiol. Mol. Biol. Rev. 2015, 79, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Marutani, Y.; Shoji, I. Cytosolic DNA-sensing immune response and viral infection. Microbiol. Immunol. 2019, 63, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors:shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B.H. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Shaw, M.H.; Kim, Y.G.; Nuñez, G. NOD-like4 receptors: Role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 2009, 4, 365–398. [Google Scholar] [CrossRef]
- Nakaya, Y.; Lilue, J.; Stavrou, S.; Moran, E.A.; Ross, S.R. AIM2-Like Receptors Positively and Negatively Regulate the Interferon Response Induced by Cytosolic DNA. MBio 2017, 8, e00944-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef]
- Shu, C.; Li, X.; Li, P. The mechanism of double-stranded DNA sensing through the cGAS-STING pathway. Cytokine Growth Factor Rev. 2014, 25, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shu, C.; Yi, G.; Chaton, C.T.; Shelton, C.L.; Diao, J.; Zuo, X.; Kao, C.C.; Herr, A.B.; Li, P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013, 39, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 2013, 9, 1003384. [Google Scholar] [CrossRef] [Green Version]
- Kalali, B.N.; Köllisch, G.; Mages, J.; Müller, T.; Bauer, S.; Wagner, H.; Ring, J.; Lang, R.; Mempel, M.; Ollert, M. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J. Immunol. 2008, 181, 2694–2704. [Google Scholar] [CrossRef] [Green Version]
- Kollisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005, 114, 531–541. [Google Scholar] [CrossRef]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Laimins, L.A. Manipulation of the innate immune response by human papillomaviruses. Virus Res. 2017, 231, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus Immune Evasion Strategies Target the Infected Cell and the Local Immune System. Front. Oncol. 2019, 9, 682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergot, A.S.; Ford, N.; Leggatt, G.R.; Wells, J.W.; Frazer, I.H.; Grimbaldeston, M.A. HPV16-E7 expression in squamous epithelium creates a local immune suppressive environment via CCL2- and CCL5-mediated recruitment of mast cells. PLoS Pathog. 2014, 10, e1004466. [Google Scholar] [CrossRef]
- James, C.D.; Fontan, C.T.; Otoa, R.; Das, D.; Prabhakar, A.T.; Wang, X.; Bristol, M.L.; Morgan, I.M. Human Papillomavirus 16 E6 and E7 Synergistically Repress Innate Immune Gene Transcription. mSphere 2020, 5, e00828-19. [Google Scholar] [CrossRef] [Green Version]
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. MBio 2016, 7, e00270-16. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Klimentová, J.; Göckel-Krzikalla, E.; Ly, R.; Gmelin, N.; Hotz-Wagenblatt, A.; Řehulková, H.; Stulík, J.; Rösl, F.; Niebler, M. Combined Transcriptome and Proteome Analysis of Immortalized Human Keratinocytes Expressing Human Papillomavirus 16 (HPV16) Oncogenes Reveals Novel Key Factors and Networks in HPV-Induced Carcinogenesis. mSphere 2019, 4, e00129-19. [Google Scholar] [CrossRef] [Green Version]
- Lo Cigno, I.; De Andrea, M.; Borgogna, C.; Albertini, S.; Landini, M.M.; Peretti, A.; Johnson, K.E.; Chandran, B.; Landolfo, S.; Gariglio, M. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J. Virol. 2015, 89, 7506–7520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccardo, E.; Lepique, A.P.; Villa, L.L. The role of inflammation in HPV carcinogenesis. Carcinogenesis 2010, 31, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Garner-Hamrick, P.A.; Fisher, C.; Lee, D.; Lambert, P.F. Methylation Patterns of Papillomaviral DNA, Its Influence on E2 Function and Implications in Viral Infection. J. Virol. 2003, 77, 12450–12459. [Google Scholar] [CrossRef] [Green Version]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [Green Version]
- Hebner, C.M.; Wilson, R.; Rader, J.; Bidder, M.; Laimins, L.A. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 2006, 87, 3183–3193. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [CrossRef]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertini, S.; Lo Cigno, I.; Calati, F.; De Andrea, M.; Borgogna, C.; Dell’Oste, V.; Landolfo, S.; Gariglio, M. HPV18 Persistence Impairs Basal and DNA Ligand-Mediated IFN-β and IFN-λ(1) Production through Transcriptional Repression of Multiple Downstream Effectors of Pattern Recognition Receptor Signaling. J. Immunol. 2018, 200, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94, e01812-19. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.; Idris, A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Pauli, E.K.; Biryukov, J.; Feister, K.F.; Meng, M.; White, E.A.; Münger, K.; Howley, P.M.; Meyers, C.; Gack, M.U. The Human Papillomavirus E6 Oncoprotein Targets USP15 and TRIM25 To Suppress RIG-I-Mediated Innate Immune Signaling. J. Virol. 2018, 92, e01737-17. [Google Scholar] [CrossRef] [Green Version]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [Green Version]
- Daud, I.I.; Scott, M.E.; Ma, Y.; Shiboski, S.; Farhat, S.; Moscicki, A.B. Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int. J. Cancer 2011, 128, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.E.; Ma, Y.; Farhat, S.; Moscicki, A.B. Expression of nucleic acid-sensing Toll-like receptors predicts HPV16 clearance associated with an E6-directed cell-mediated response. Int. J. Cancer 2015, 136, 2402–2408. [Google Scholar] [CrossRef] [Green Version]
- Halec, G.; Scott, M.E.; Farhat, S.; Darragh, T.M.; Moscicki, A.B. Toll-like receptors: Important immune checkpoints in the regression of cervical intra-epithelial neoplasia 2. Int. J. Cancer 2018, 143, 2884–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Pépin, G.; Gantier, M.P. cGAS-STING Activation in the Tumor Microenvironment and Its Role in Cancer Immunity. Adv. Exp. Med. Biol. 2017, 1024, 175–194. [Google Scholar] [CrossRef]
- Diner, B.A.; Cristea, I.M. Blowing Off Steam: Virus Inhibition of cGAS DNA Sensing. Cell Host Microbe 2015, 18, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Hopcraft, S.E.; Damania, B. Tumour viruses and innate immunity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160267. [Google Scholar] [CrossRef] [PubMed]
- Bussey, K.A.; Brinkmann, M.M. Strategies for immune evasion by human tumor viruses. Curr. Opin. Virol. 2018, 32, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.D. Nucleotide sequences and further characterization of human papillomavirus DNA present in the CaSki, SiHa and HeLa cervical carcinoma cell lines. J. Gen. Virol. 1999, 80, 1725–1733. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Wen, H.; Ting, J.P. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy 2013, 9, 432–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kansy, B.A.; Li, J.; Cong, L.; Liu, Y.; Trivedi, S.; Wen, H.; Ting, J.P.; Ouyang, H.; Ferris, R.L. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene 2016, 35, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Zhang, Y.; Dittmer, D.P.; Mieczkowski, P.A.; Host, K.M.; Fusco, W.G.; Duncan, J.A.; Damania, B. RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. MBio 2018, 9, e00823-18. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.J.; Davis, M.E.; Gack, M.U. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. 2014, 25, 491–505. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef]
- Andersen, J.M.; Al-Khairy, D.; Ingalls, R.R. Innate immunity at the mucosal surface: Role of toll-like receptor 3 and toll-like receptor 9 in cervical epithelial cell responses to microbial pathogens. Biol. Reprod. 2006, 74, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Anwar, M.A.; Park, S.; Jafri, S.S.; Choi, S. In silico mechanistic analysis of IRF3 inactivation and high-risk HPV E6 species-dependent drug response. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Bristol, M.L.; Das, D.; Morgan, I.M. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses 2017, 9, 268. [Google Scholar] [CrossRef] [Green Version]
- Wallace, N.A. Catching HPV in the Homologous Recombination Cookie Jar. Trends Microbiol. 2020, 28, 191–201. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Hoffmann, J.A.; Galluzzi, L. Pharmacological modulation of nucleic acid sensors—Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2019, 18, 845–867. [Google Scholar] [CrossRef]
- Goulet, M.L.; Olagnier, D.; Xu, Z.; Paz, S.; Belgnaoui, S.M.; Lafferty, E.I.; Janelle, V.; Arguello, M.; Paquet, M.; Ghneim, K.; et al. Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog. 2013, 9, e1003298. [Google Scholar] [CrossRef]
- Beljanski, V.; Chiang, C.; Kirchenbaum, G.A.; Olagnier, D.; Bloom, C.E.; Wong, T.; Haddad, E.K.; Trautmann, L.; Ross, T.M.; Hiscott, J. Enhanced Influenza Virus-Like Particle Vaccination with a Structurally Optimized RIG-I Agonist as Adjuvant. J. Virol. 2015, 89, 10612–10624. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Beljanski, V.; Yin, K.; Olagnier, D.; Ben Yebdri, F.; Steel, C.; Goulet, M.L.; DeFilippis, V.R.; Streblow, D.N.; Haddad, E.K.; et al. Sequence-Specific Modifications Enhance the Broad-Spectrum Antiviral Response Activated by RIG-I Agonists. J. Virol. 2015, 89, 8011–8025. [Google Scholar] [CrossRef] [Green Version]
- Castiello, L.; Zevini, A.; Vulpis, E.; Muscolini, M.; Ferrari, M.; Palermo, E.; Peruzzi, G.; Krapp, C.; Jakobsen, M.; Olagnier, D.; et al. An optimized retinoic acid-inducible gene I agonist M8 induces immunogenic cell death markers in human cancer cells and dendritic cell activation. Cancer Immunol. Immunother. 2019, 68, 1479–1492. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mechanism or Target | Cellular Model | Reference |
|---|---|---|
| cGAS/STING/TBK1 signaling pathway | ||
| HPV18 E7 binds and antagonizes STING. | HeLa cells and mouse embryo fibroblasts stably transduced with HPV18 E6 or E7 expressing retroviruses. | Lau, L. et al., 2015 [94] |
| Epigenetic silencing of cGAS and STING genes through HPV16/18 E7-mediated induction of the methyltransferase SUV39H1. | HeLa, CaSki, NIKSmcHPV18 and HEK 293 cells expressing either HPV16 or HPV18 E6 and E7. | Albertini, S. et al., 2018 [95]; Lo Cigno, I. et al., 2020 [96] |
| HPV16 E7 hijacks NLRX1 to induce STING degradation via an autophagy-dependent mechanism. | HNSCC-derived cell lines ectopically expressing HPV16 E7 and a syngeneic C57/BL/6 model of HPV+ HNSCC. | Luo, X. et al., 2019 [97] |
| The H3K4 lysine demethylases KDM5B and KDM5C epigenetically suppress STING mRNA expression levels. | Breast cancer cells, HPV+ head and neck and cervical carcinomas. | Wu, L. et al., 2018 [98] |
| Impaired IFNβ gene transcriptional activation upon stimulation with STING agonists. | HPV16+ HNSCC-derived cell lines. | Shaikh, M.H. et al., 2019 [99] |
| RIG-I/MAVS/TBK1 signaling pathway | ||
| HPV16 E6 forms a ternary E6-TRIM25-USP15 complex that reduces TRIM25 protein-stability, leading to reduced ubiquitination of RIG-I and suppression of its ability to interact with MAVS. | HEK 293T and the cervical carcinoma-derived cell line C33a ectopically expressing FLAG-tagged E6 of HPV16. | Chiang, C. et al., 2018 [100] |
| HPV16/18 E7 induces the transcription of SUV39H1, which promotes epigenetic silencing of RIG-I. | HeLa, CaSki, NIKSmcHPV18 and HEK 293 cells expressing either HPV16/18 E6 or E7. | Albertini, S. et al., 2018 [95]; Lo Cigno, I. et al., 2020 [96] |
| TLR signaling pathway | ||
| HPV16 E6/E7 induce downregulation of TLR9 expression at both mRNA and protein levels. | Human primary keratinocytes stably transduced with HPV16E6/E7 expressing retroviruses, HeLa, SiHa and CaSki. | Hasan, U.A. et al., 2007 [101] |
| HPV16 suppresses TLR expression in the cervical mucosa, contributing to viral persistence. | Cervical cytobrush samples. | Daud, I.I. et al., 2011 [102]; Scott, M.E. et al., 2015 [103]; Halec, G. et al., 2018 [104] |
| PRR-Related Transcription Factors | ||
| HPV16 E6 binds IRF3 and impairs its transcriptional activity. | In vitro synthesized protein, and HPV16 E6-transfected cells, including primary human keratinocytes. | Ronco, L.V. et al., 1998 [93] |
| HPV16 E7 binds IRF1 and impairs its DNA binding and transcriptional activity. | In vitro synthesized HPV16 E7 and HPV16E7-transfected cells. | Park, J.S. et al., 2000 [90]; Perea, S.E. et al., 2000 [91]; Um, S.J. et al., 2002 [92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Cigno, I.; Calati, F.; Albertini, S.; Gariglio, M. Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins. Pathogens 2020, 9, 292. https://doi.org/10.3390/pathogens9040292
Lo Cigno I, Calati F, Albertini S, Gariglio M. Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins. Pathogens. 2020; 9(4):292. https://doi.org/10.3390/pathogens9040292
Chicago/Turabian StyleLo Cigno, Irene, Federica Calati, Silvia Albertini, and Marisa Gariglio. 2020. "Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins" Pathogens 9, no. 4: 292. https://doi.org/10.3390/pathogens9040292
APA StyleLo Cigno, I., Calati, F., Albertini, S., & Gariglio, M. (2020). Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins. Pathogens, 9(4), 292. https://doi.org/10.3390/pathogens9040292