Effects of Mycoplasmas on the Host Cell Signaling Pathways


 ,
, 
{kind=link}
Abstract
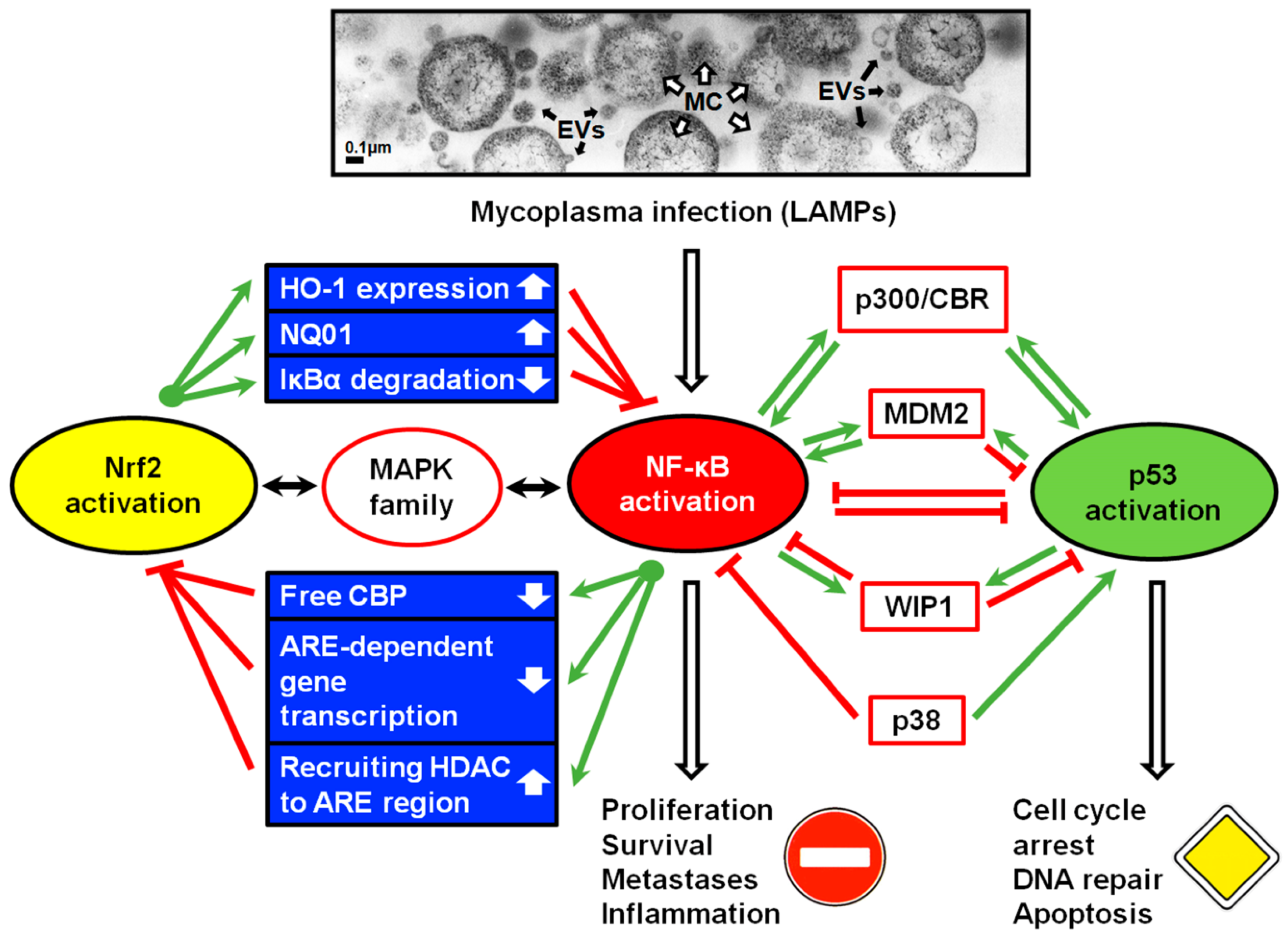
:1. Introduction
2. Mycoplasmas Modulate Inflammatory Response
3. Mycoplasmal Infections Promote Tumor Transformation
4. NF-κB and p53 Pathways in Eukaryotic Cells
5. Molecular Mechanisms of p53 and NF-κB Antagonism
5.1. The Contest for p300 Acetyltransferase
5.2. p53- and NF-κB- Targeting Micro-RNAs
6. Effects of Mycoplasmas on Expression of Micro-RNAs
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Browning, G.; Citti, C. Mollicutes: Molecular Biology and Pathogenesis; Caister Academic Press: Norfolk, UK, 2014; p. 350. [Google Scholar]
- Borchsenius, S.N.; Chernova, O.A.; Chernov, V.M.; Vishnyakov, I.E. Mikoplazmy v Biologii i Meditsine Nachala 21 Veka (Mycoplasmas in Biology and Medicine of the Early 21st Century); Nauka: St. Petersburg, Russia, 2016; p. 334. [Google Scholar]
- Razin, S.; Hayflick, L. Highlights of mycoplasma research - an historical perspective. Biologicals 2010, 38, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Rottem, S.; Kosower, N.S.; Kornspan, J.D. Contamination of tissue cultures by mycoplasmas. In Biomedical Tissue Culture; Ceccherini-Nelli, L., Matteoli, B., Eds.; IntecOpen: London, UK, 2012; pp. 35–38. [Google Scholar] [CrossRef] [Green Version]
- Uphoff, C.C.; Drexler, H.G. Detecting mycoplasma contamination in cell cultures by polymerase chain reaction. Methods Mol. Biol. 2011, 731, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Yavlovich, A.; Tarshis, M.; Rottem, S. Internalization and intracellular survival of Mycoplasma pneumoniae by non-phagocytic cells. FEMS Microbiol. Lett. 2004, 233, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Paessler, M.; Levinson, A.; Patel, J.B.; Schuster, M.; Minda, M.; Nachamkin, I. Disseminated Mycoplasma orale infection in a patient with common variable immunodeficiency syndrome. Diagn. Microbiol. Infect. Dis. 2002, 44, 201–204. [Google Scholar] [CrossRef]
- Morozova, A.V.; Borchsenius, S.N.; Vishnyakov, I.E.; Malinin, A.Y. Testing the purity of cell cultures using clinical diagnostic PCR kits. Cell Tiss. Biol. 2017, 11, 250–259. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Detection of mycoplasma contaminations. Methods Mol. Biol. 2013, 946, 1–13. [Google Scholar] [CrossRef]
- Feng, S.H.; Tsai, S.; Rodriguez, J.; Lo, S.C. Mycoplasmal infections prevent apoptosis and induce malignant transformation of interleukin-3-dependent 32D hematopoietic cells. Mol. Cell. Biol. 1999, 19, 7995–8002. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; Wear, D.J.; Shih, J.W.; Lo, S.C. Mycoplasmas and oncogenesis: Persistent infection and multistage malignant transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 10197–10201. [Google Scholar] [CrossRef] [Green Version]
- Grace, J.T.; Horoszewicz, J.S.; Stim, T.B.; Mirand, E.A.; James, C. Mycoplasmas (PPLO) and human leukemia and lymphoma. Cancer 1965, 18, 1369–1376. [Google Scholar] [CrossRef]
- Zhang, B.; Shih, J.W.; Wear, D.J.; Tsai, S.; Lo, S.C. High-level expression of H-ras and c-myc oncogenes in mycoplasma-mediated malignant cell transformation. Proc. Soc. Exp. Biol. Med. 1997, 214, 359–366. [Google Scholar] [CrossRef]
- Razin, S.; Yogev, D.; Naot, Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 1998, 62, 1094–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, M.A.; Frasca, S.; Rood, D.; Cecchini, K.; Gladd, M.; Geary, S.J.; Silbart, L.K. Correlates of immune protection in chickens vaccinated with Mycoplasma gallisepticum strain GT5 following challenge with pathogenic M. gallisepticum strain R(low). Infect. Immun. 2005, 73, 5410–5419. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Loveless, R.W.; Griffiths, S.; Fryer, P.R.; Blauth, C.; Feizi, T. Immunoelectron microscopic studies reveal differences in distribution of sialo-oligosaccharide receptors for Mycoplasma pneumoniae on the epithelium of human and hamster bronchi. Infect Immun. 1992, 60, 4015–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Yerneni, L.K. Semi-automated relative quantification of cell culture contamination with mycoplasma by Photoshop-based image analysis on immunofluorescence preparations. Biologicals 2009, 37, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Peltier, M.R.; Freeman, A.J.; Mu, H.H.; Cole, B.C. Characterization of the macrophage-stimulating activity from Ureaplasma urealyticum. Am. J. Reprod. Immunol. 2007, 57, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Takeuchi, O.; Akira, S. Recognition of lipopeptides by Toll-like receptors. J. Endotoxin Res. 2002, 8, 459–463. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kaufmann, A.; Grote, K.; Kawai, T.; Hoshino, K.; Morr, M.; Mühlradt, P.F.; Akira, S. Cutting edge: Preferentially the R-stereoisomer of the mycoplasma lipopeptide macrophage-activating lipoptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 2000, 164, 554–557. [Google Scholar] [CrossRef] [Green Version]
- Okusawa, T.; Fujita, M.; Nakamura, J.; Into, T.; Yasuda, M.; Yoshimura, A.; Hara, Y.; Hasebe, A.; Golenbock, D.T.; Morita, M.; et al. Relationship between structures and biological activities of mycoplasmal diacylated lipopeptides and their recognition by Toll-like receptors 2 and 6. Infect. Immun. 2004, 72, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Kida, Y.; Kuwano, K. Triacylated lipoproteins derived from Mycoplasma pneumoniae activate nuclear factor-kappaB through Toll-like receptors 1 and 2. Immunology 2007, 121, 473–483. [Google Scholar] [CrossRef]
- Yong, Y.; Liu, S.; Hua, G.; Jia, R.; Zhao, Y.; Sun, X.; Liao, M.; Ju, X. Identification and functional characterization of Toll-like receptor 2-1 in geese. BMC Vet. Res. 2015, 11, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, A.; Mühlradt, P.F.; Gemsa, D.; Sprenger, H. Induction of cytokines and chemokines in human monocytes by Mycoplasma fermentans - derived lipoprotein MALP-2. Infect. Immun. 1999, 67, 6303–6308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, J.; Frasca, S., Jr.; Cecchini, K.; Rood, D.; Nyaoke, A.C.; Geary, S.J.; Silbart, L.K. Chemokine and cytokine gene expression profiles in chickens inoculated with Mycoplasma gallisepticum strains Rlow or GT5. Vaccine 2007, 25, 8611–8621. [Google Scholar] [CrossRef] [PubMed]
- Deiters, U.; Mühlradt, P.F. Mycoplasmal lipopeptide MALP-2 induces the chemoattractant proteins macrophage inflammatory protein 1α (MIP-1α), monocyte chemoattractant protein 1, and MIP-2 and promotes leukocyte infiltration in mice. Infect. Immun. 1999, 67, 3390–3398. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Zhao, C.; Hu, Q.; Sun, J.; Peng, X. Roles of Toll-like receptors 2 and 6 in the inflammatory response to Mycoplasma gallisepticum infection in DF-1 cells and in chicken embryos. Dev. Comp. Immunol. 2016, 59, 39–47. [Google Scholar] [CrossRef]
- Shimizu, T.; Kida, Y.; Kuwano, K. Mycoplasma pneumoniae-derived lipopeptides induce acute inflammatory responses in the lungs of mice. Infect. Immun. 2008, 76, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.; Lichtman, A. Innate Immunity. The Early Defence Against Infections. In Basic Immunology. Functions and Disorders of the Immune System, 3rd ed.; Saunders (Elsevier): Philadelphia, PA, USA, 2009; pp. 23–44. [Google Scholar]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Prawan, A.; Saw, C.L.; Khor, T.O.; Keum, Y.S.; Yu, S.; Hu, L.; Kong, A.N. Anti-NF-kappaB and antiinlammatory activities of synthetic isothiocyanates: Effect of chemical structures and cellular signaling. Chem. Biol. Interact. 2009, 179, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Vasudevan, P.; Zhong, L.; Kim, G.; Samant, S.; Parekh, V.; Pillai, V.B.; Ravindra, P.V.; Gupta, M.; Jeevanandam, V.; et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 2012, 18, 1643–1650. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Lee, S.; Gugiu, B.G.; Koroniak, L.; Jung, M.E.; Berliner, J.; Cheng, J.; Li, R. Fatty acid epoxyisoprostane E2 stimulates an oxidative stress response in endothelial cells. Biochim. Biophys. Res. Commun. 2014, 444, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.; Chen, X.; Mackman, N.; Ogborne, R.; O’Connell, M. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408–4415. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Saidu, N.E.; Touma, R.; Asali, I.A.; Jacob, C.; Montenarh, M. Diallyl tetrasulfane activates both the eIF2alpha and Nrf2/HO-1 pathways. Biochim. Biophys. Acta 2013, 1830, 2214–2225. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2–ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Luo, L.; Namani, A.; Wang, X.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; You, X.; Zeng, Y.; He, J.; Liu, L.; Deng, Z.; Jiang, C.; Wu, H.; Zhu, C.; Yu, M.; et al. Mycoplasma fermentans MALP-2 induces heme oxygenase-1 expression via mitogen-activated protein kinases and Nrf2 pathways to modulate cyclooxygenase 2 expression in human monocytes. Clin. Vaccine Immunol. 2013, 20, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Zappulla, F.; Silbart, L.K. Mycoplasma gallisepticum lipid associated membrane proteins up-regulate inflammatory genes in chicken tracheal epithelial cells via TLR-2 ligation through an NF-κB dependent pathway. PLoS ONE 2014, 9, e112796. [Google Scholar] [CrossRef]
- He, L.; You, X.; Li, G.; Zeng, Y.; Li, R.; Zhu, C.; Yu, M.; Wu, Y. Mycoplasma genitalium-derived lipid associated membrane proteins negatively regulate cytokine secretion by inducing HO-1 expression in placental trophoblast cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 194–198. [Google Scholar]
- Hu, J.; Chen, C.; Ou, G.; You, X.; Tan, T.; Hu, X.; Zeng, Y.; Yu, M.; Zhu, C. Nrf2 regulates the inflammatory response, including heme oxygenase-1 induction, by Mycoplasma pneumoniae lipid-associated membrane proteins in THP-1 cells. Pathog. Dis. 2017, 75, ftx044. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Kida, Y.; Kuwano, K. Lipid-associated membrane proteins of Mycoplasma fermentans and M. penetrans activate human immunodeficiency virus long-terminal repeats through Toll-like receptors. Immunology 2004, 113, 121–129. [Google Scholar] [CrossRef]
- Soares, M.P.; Bach, F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Weng, C.; Wang, Y.; Wu, M. Lipoic acid ameliorates arsenic trioxide-induced HO-1 expression and oxidative stress in THP-1 monocytes and macrophages. Chem. Biol. Interact. 2011, 190, 129–138. [Google Scholar] [CrossRef]
- Liang, C.; Xue, Z.; Cang, J.; Wang, H.; Li, P. Dimethyl sulfoxide induces heme oxygenase-1 expression via JNKs and Nrf2 pathways in human umbilical vein endothelial cells. Mol. Cell. Biochem. 2011, 355, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.Y.; Yeh, W.L.; Huang, S.M.; Tang, C.H.; Lin, H.Y.; Chou, S.J. Osteopontin increases heme oxygenase-1 expression and subsequently induces cell migration and invasion in glioma cells. Neuro. Oncol. 2012, 14, 1367–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Li, H.; Cao, F.; Zhen, L.; Bai, J.; Yuan, S.; Mei, Y. 1,2,3,4,6-penta-O-galloyl-beta-d-glucose protects PC12 cells from MPP(+)-mediated cell death by inducing heme oxygenase-1 in an ERK- and Akt-dependent manner. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 737–745. [Google Scholar] [CrossRef]
- Ishfaq, M.; Chen, C.; Bao, J.; Zhang, W.; Wu, Z.; Wang, J.; Liu, Y.; Tian, E.; Hamid, S.; Li, R.; et al. Baicalin ameliorates oxidative stress and apoptosis by restoring mitochondrial dynamics in the spleen of chickens via the opposite modulation of NF-κB and Nrf2/HO-1 signaling pathway during Mycoplasma gallisepticum infection. Poult. Sci. 2019, 98, 6296–6310. [Google Scholar] [CrossRef]
- Li, J.; Qiao, Z.; Hu, W.; Zhang, W.; Shah, S.W.A.; Ishfaq, M. Baicalin mitigated Mycoplasma gallisepticum-induced structural damage and attenuated oxidative stress and apoptosis in chicken thymus through the Nrf2/HO-1 defence pathway. Vet. Res. 2019, 50, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimolai, N. Do mycoplasmas cause human cancer? Can. J. Microbiol. 2001, 47, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Polianskaia, G.G.; Efremova, T.E. Effect of mycoplasma contamination of a human cervical carcinoma cell line M HeLa clone 11 on karyotypic variability. Tsitologiia 2000, 42, 794–801. [Google Scholar]
- Poljanskaya, G.G.; Efremova, T.N. Effect of Mycoplasma salivarium with and without L-arginine on karyotypic variability in cell line of Indian muntjac skin fibroblasts at long-term cultivation. Cell. Tiss. Biol. 2011, 5, 54–61. [Google Scholar] [CrossRef]
- Hopfe, M.; Deenen, R.; Degrandi, D.; Koher, K.; Henrich, B. Host cell response to persistent mycoplasmas—Different stages in infection of HeLa cells with Mycoplasma hominis. PLoS ONE 2013, 8, e54219. [Google Scholar] [CrossRef]
- Mou, H.Q.; Lu, J.; Zhu, S.F.; Lin, C.L.; Tian, G.Z.; Xu, X.; Zhao, W.J. Transcriptomic analysis of paulownia infected by paulownia witches’-broom phytoplasma. PLoS ONE 2013, 8, e77217. [Google Scholar] [CrossRef]
- Lavrič, M.; Maughan, M.N.; Bliss, T.W.; Dohms, J.E.; Bencina, D.; Keeler, C.L., Jr.; Narat, M. Gene expression modulation in chicken macrophages exposed to Mycoplasma synoviae or Escherichia coli. Vet. Microbiol. 2008, 126, 111–121. [Google Scholar] [CrossRef]
- Zhang, S.; Wear, D.J.; Lo, S. Mycoplasmal infections alter gene expression in cultured human prostatic and cervical epithelial cells. FEMS Immunol. Med. Microbiol. 2000, 27, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Namiki, K.; Goodison, S.; Porvasnik, S.; Allan, R.W.; Iczkowski, K.A.; Urbanek, C.; Reyes, L.; Sakamoto, N.; Rosser, C.J. Persistent exposure to mycoplasma induces malignant transformation of human prostate cells. PLoS ONE 2009, 4, e6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oriel, J.D. Role of genital mycoplasmas in nongonococcal urethritis and prostatitis. Sex Transm. Dis. 1983, 10, 263–270. [Google Scholar]
- Barykova, Y.A.; Logunov, D.Y.; Shmarov, M.M.; Vinarov, A.Z.; Fiev, D.N.; Vinarova, N.A.; Rakovskaya, I.V.; Baker, P.S.; Shyshynova, I.; Stephenson, A.J.; et al. Association of Mycoplasma hominis infection with prostate cancer. Oncotarget 2011, 2, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.E.; Agarwal, S.; Kestler, D.P. Induction of leukemia cell differentiation and apoptosis by recombinant P48, a modulin derived from Mycoplasma fermentans. Biochem. Biophys. Res. Commun. 2000, 269, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, I.A.; Vaughan, A.T.; Khodarev, N.N. Mycoplasma infection can sensitize host cells to apoptosis through contribution of apoptotic-like endonuclease(s). Immunol. Cell. Biol. 1998, 76, 526–534. [Google Scholar] [CrossRef]
- Zhang, S.; Lo, S.C. Effect of mycoplasmas on apoptosis of 32D cells is species-dependent. Curr. Microbiol. 2007, 54, 388–395. [Google Scholar] [CrossRef]
- Ak, P.; Levine, A.J. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef]
- Chiao, P.J.; Miyamoto, S.; Verma, I.M. Autoregulation of I kappa B alpha activity. Proc. Natl. Acad. Sci. USA 1994, 91, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Lee, T.L.; Yang, X.P.; Yan, B.; Friedman, J.; Duggal, P.; Bagain, L.; Dong, G.; Yeh, N.T.; Wang, J.; Zhou, J.; et al. A novel nuclear factor-kappaB gene signature is differentially expressed in head and neck squamous cell carcinomas in association with TP53 status. Clin. Cancer Res. 2007, 13, 5680–5691. [Google Scholar] [CrossRef] [Green Version]
- Gurova, K.V.; Hill, J.E.; Guo, C.; Prokvolit, A.; Burdelya, L.G.; Samoylova, E.; Khodyakova, A.V.; Ganapathi, R.; Ganapathi, M.; Tararova, N.D.; et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-κB-dependent mechanism of p53 suppression in tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 17448–17453. [Google Scholar] [CrossRef] [Green Version]
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. Lysine-specific modifications of p53: A matter of life and death? Oncotarget 2013, 4, 1556–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudkov, A.V.; Komarova, E.A. p53 and the carcinogenicity of chronic inflammation. Cold Spring Harb. Perspect. Med. 2016, 6, a026161. [Google Scholar] [CrossRef] [Green Version]
- Poyurovsky, M.V.; Prives, C. Unleashing the power of p53: Lessons from mice and men. Genes Dev. 2006, 20, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Banks, L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 2001, 20, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, K.; Witkowski, J.M. Viral strategies in modulation of NF-kappaB activity. Arch. Immunol. Ther. Exp. (Warsz.) 2003, 51, 367–376. [Google Scholar] [PubMed]
- Logunov, D.Y.; Scheblyakov, D.V.; Zubkova, O.V.; Shmarov, M.M.; Rakovskaya, I.V.; Gurova, K.V.; Gudkov, A.V. Mycoplasma infection suppresses p53, activates NF-κB and cooperates with oncogenic Ras in rodent fibroblast transformation. Oncogene 2008, 27, 4521–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szołtysek, K.; Pietranek, K.; Kalinowska-Herok, M.; Pietrowska, M.; Kimmel, M.; Widłak, P. TNFalpha-induced activation of NFkappaB protects against UV-induced apoptosis specifically in p53-proficient cells. Acta Biochim. Pol. 2008, 55, 741–748. [Google Scholar] [CrossRef]
- Weisz, L.; Damalas, A.; Liontos, M.; Karakaidos, P.; Fontemaggi, G.; Maor-Aloni, R.; Kalis, M.; Levrero, M.; Strano, S.; Gorgoulis, V.G.; et al. Mutant p53 enhances nuclear factor κB activation by tumor necrosis factor α in cancer cells. Cancer Res. 2007, 67, 2396–2401. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, D.; Cogswell, P.; Baldwin, A.S. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006, 20, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, L.J.; Eckmann, L.; Greten, F.R.; Chae, S.; Li, Z.W.; Myhre, G.M.; Robine, S.; Karin, M.; Kagnoff, M.F. IκB-kinaseβ-dependent NF-κB activation provides radioprotection to the intestinal epithelium. Proc. Natl. Acad. Sci. USA 2004, 101, 2452–2457. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-κB and p53. Mol. Cell Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; van Hensbergen, Y.; Bedi, G.C.; Giordano, A.; El-Deiry, W.S.; Fuchs, E.J.; Bedi, A. p53-mediated repression of nuclear factor-kappaB RelA via the transcriptional integrator p300. Cancer Res. 1998, 58, 4531–4536. [Google Scholar] [PubMed]
- Ea, C.K.; Baltimore, D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. USA 2009, 106, 18972–18977. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.D.; Huang, B.; Li, M.; Lamb, A.; Kelleher, N.L.; Chen, L.F. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009, 28, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-κB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Barlev, N.A.; Sayan, B.S.; Candi, E.; Okorokov, A.L. The microRNA and p53 families join forces against cancer. Cell Death Differ. 2010, 17, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Farazi, T.A.; Hoell, J.I.; Morozov, P.; Tuschl, T. MicroRNAs in human cancer. Adv. Exp. Med. Biol. 2013, 774, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Ghose, J.; Sinha, M.; Das, E.; Jana, N.R.; Bhattacharyya, N.P. Regulation of miR-146a by RelA/NFkB and p53 in STHdhQ111/HdhQ111 cells, a cell model of Huntington’s disease. PLoS ONE 2011, 6, e23837. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Niu, J.; Shi, Y.; Ouyang, H.; Wu, Z.H. NF-κB-dependent microRNA-125b up-regulation promotes cell survival by targeting p38α upon ultraviolet radiation. J. Biol. Chem. 2012, 287, 33036–33047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Luo, F.; Liu, X.; Lu, L.; Xu, H.; Yang, Q.; Xue, J.; Shi, L.; Li, J.; Zhang, A.; et al. NF-κB-regulated exosomal miR-155 promotes the inflammation associated with arsenite carcinogenesis. Cancer Lett. 2017, 388, 21–33. [Google Scholar] [CrossRef]
- Moore, R.; Ooi, H.K.; Kang, T.; Bleris, L.; Ma, L. MiR-192-mediated positive feedback loop controls the robustness of stress - induced p53 oscillations in breast cancer cells. PLoS Comput. Biol. 2015, 11, e1004653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, E.; Cai, G.; Kuehn, L.A.; Register, K.B.; McDaneld, T.G.; Neill, J.D. Association of microRNAs with antibody response to Mycoplasma bovis in beef cattle. PLoS ONE 2016, 11, e0161651. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Bi, D.; Hou, Y.; Zhao, Y.; Sun, J.; Peng, X. gga-miR-101-3p plays a key role in Mycoplasma gallisepticum (HS strain) infection of chicken. Int. J. Mol. Sci. 2015, 16, 28669–28682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wang, Z.; Hou, Y.; Zhang, K.; Peng, X. gga-miR-99a targets SMARCA5 to regulate Mycoplasma gallisepticum (HS strain) infection by depressing cell proliferation in chicken. Gene 2017, 627, 239–247. [Google Scholar] [CrossRef]
- Mueller, A.C.; Sun, D.; Dutta, A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene 2013, 32, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Han, Y.; Zhao, Y.; Sun, Y.; Zou, M.; Fu, Y.; Peng, X. Upregulated gga-miR-16-5p inhibits the proliferation cycle and promotes the apoptosis of MG-infected DF-1 cells by repressing PIK3R1-mediated the PI3K/Akt/NF-κB pathway to exert anti-inflammatory effect. Int. J. Mol. Sci. 2019, 20, 1036. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Zou, M.; Zhao, Y.; Zhang, K.; Sun, Y.; Peng, X. Up-regulation of miR-130b-3p activates the PTEN/PI3K/AKT/NF-κB pathway to defense against Mycoplasma gallisepticum (HS strain) infection of chicken. Int. J. Mol. Sci. 2018, 19, 2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Um, J.H.; Cho, J.H.; Lee, H.J. Tiny RNAs and their voyage via extracellular vesicles: Secretion of bacterial small RNA and eukaryotic microRNA. Exp. Biol. Med. 2017, 242, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Chernov, V.M.; Chernova, O.A.; Mouzykantov, A.A.; Medvedeva, E.S.; Baranova, N.B.; Malygina, T.Y.; Aminov, R.I.; Trushin, M.V. Antimicrobial resistance in mollicutes: Known and newly emerging mechanisms. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Xu, X.; Wang, Y.; Shen, X.; Chen, Z.; Yang, J. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infect. Immun. 2008, 76, 4405–4413. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borchsenius, S.N.; Vishnyakov, I.E.; Chernova, O.A.; Chernov, V.M.; Barlev, N.A. Effects of Mycoplasmas on the Host Cell Signaling Pathways. Pathogens 2020, 9, 308. https://doi.org/10.3390/pathogens9040308
Borchsenius SN, Vishnyakov IE, Chernova OA, Chernov VM, Barlev NA. Effects of Mycoplasmas on the Host Cell Signaling Pathways. Pathogens. 2020; 9(4):308. https://doi.org/10.3390/pathogens9040308
Chicago/Turabian StyleBorchsenius, Sergei N., Innokentii E. Vishnyakov, Olga A. Chernova, Vladislav M. Chernov, and Nikolai A. Barlev. 2020. "Effects of Mycoplasmas on the Host Cell Signaling Pathways" Pathogens 9, no. 4: 308. https://doi.org/10.3390/pathogens9040308
APA StyleBorchsenius, S. N., Vishnyakov, I. E., Chernova, O. A., Chernov, V. M., & Barlev, N. A. (2020). Effects of Mycoplasmas on the Host Cell Signaling Pathways. Pathogens, 9(4), 308. https://doi.org/10.3390/pathogens9040308