1. Introduction
Upon sensing virus entry through families of pathogen recognition receptors (PRRs), host cells unleash a wide range of signals, which culminate in the initiation of anti-viral machinery. The induction of type I interferons (IFNs), including IFN-αs and IFN-β, type III IFNs (IFN-λs), and prototypical proinflammatory cytokines, including interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α) are among the hallmarks of early host responses against invading viral pathogens. The nuclear factor kappa B (NF-κB) complex plays a key role in linking the presence of danger signals to the initiation of both inflammatory response and cellular anti-viral status [
1,
2]. Cellular localization of NF-κB depends on the activity of the inhibitor of kappa B kinase (IKK) complex. The IKK complex consists of two kinase subunits, IKKα (IKBKA) and IKKβ (IKBKB), and a regulatory subunit IKKγ/NF-κB essential modifier (NEMO). The IKBKB and NEMO are important for canonical NF-κB pathway regulation [
3]. Phosphorylation of the inhibitor of NF-κB (IκB) by the IKK complex is required to allow NF-κB to be released from IκB and translocated into the nucleus to regulate the transcription of genes participating in host immune and inflammatory responses [
4,
5].
Porcine epidemic diarrhea virus (PEDV) is the major causative agent of severe watery diarrhea with very high mortality in neonatal piglets [
6]. PEDV belongs to the
Alphacoronavirus genus in the
Coronaviridae family. Its positive-sense single-stranded RNA of approximately 28 kb in length encodes viral replicase polyprotein (pp) 1a and pp1ab; structural proteins: spike(S), envelope (E), membrane (M), and the nucleocapsid (N) proteins; and an accessory protein ORF3. We previously employed a proteomic-based approach to identify cellular proteins that potentially interact with the PEDV ORF3 protein [
7]. Among many candidates, IKKβ, or IKBKB (IKK2), was identified as one of the ORF3 interacting partners [
7]. Providing that other accessory proteins of other coronaviruses [
8,
9] have been shown to interfere with type I IFN and proinflammatory cytokine productions, it is of interest to examine whether PEDV ORF3 could likewise modulate immune signaling pathways especially through the IKBKB interaction.
In this study, we investigated the potential roles of PEDV ORF3 in the host immune modulation. In particular, we hypothesized that ORF3 directly interacts with IKBKB, leading to the dysregulation of the NF-κB-mediated activity such as type I interferon (IFN) and proinflammatory cytokine productions. We demonstrated here that IKBKB-mediated NF-κB and IFN-β promoter activities were differently affected by the presence of ORF3. Our data not only underscore the role of ORF3 in manipulating the host’s immune response via IKBKB, but also provide further insights into our understanding of PEDV and host interaction.
3. Discussion
Innate immune response is typically activated soon after viral infection. However, many viruses have evolved mechanisms to antagonize host innate immune functions for the purpose of facilitating their efficient replication and infection. PEDV utilizes its many proteins to restrict a host antiviral state. For example, N protein was identified as an interferon (IFN) antagonist by interfering with interferon regulatory factor 3 (IRF3) and TANK-binding kinase 1 (TBK1) interaction and consequently inhibited IFN production [
16]. Non-structural protein 5 (nsp5) or 3C-like protease disrupted RIG-I/melanoma differentiation-associated gene 5 (MDA5) signaling by cleaving NEMO, resulting in an inhibition of IFN induction [
19]. PEDV nonstructural protein 1 (nsp1) suppressed IRF1-mediated IFN-λ response [
20]. While very little is known about the role of PEDV sole accessory protein as a potential host immune response antagonist, ORF3 has been proposed as a virulent factor acting to subdue NF-κB and IRF1 promoter activation [
17,
20].
Our proteomics analysis of ORF3 interactome identified IKBKB and other cellular proteins involved in host immune responses [
7]. As IKBKB acts as a central regulator in controlling key immune signaling pathways, it is of interest to initially examine whether ORF3 could interfere with the immune signaling pathway through IKBKB binding. IKBKB is a component of the IKK kinase complexes, which comprise two kinases (IKBKA and IKBKB) and a regulatory subunit, NEMO/IKKγ. Phosphorylation of IkBα by IKK complexes is required to break up the binding between IkBα and NF-κB, and subsequently to release NF-κB into the nucleus. Nuclear NF-κB binds to NF- κB promoter to regulate transcription of many genes participating in host immune and inflammatory responses [
4,
21]. In our study, we speculated that IKBKB could be one of the key host factors involved in restricting PEDV replication in VeroE6 cells. Although PEDV
AV12-ORF3 replication was recovered in IKBKB-knock out cells, we cannot rule out the possibility that other PEDV proteins may also play a role in the reconstituted ability to replicate [
16,
19]. Vero cells are type I interferon-deficient [
22,
23], and thereby they are not responsive to IKBKB-induced type I IFN induction. However, in addition to regulation of type I IFN production, IKBKB as a master regulator of innate immunity takes part in various immune signaling pathways and other cellular mechanisms. Importantly, IKBKB has a direct role in the NF-кB signaling pathway which is essential for proinflammatory cytokine production and autophagy during virus infection [
24]. For PEDV, the virus inhibited the induction of TNF-α in Vero and MARC-145 cells. There are several viral proteins identified to regulate NF-кB signaling pathway. For instance, nsp1 has been demonstrated to inhibit NF-κB-mediated proinflammatory cytokines, resulting in supporting virus propagation at an early time point [
17]. We thus speculated that the overexpression of IKBKB mediates NF-кB activation and proinflammatory cytokine production to counteract with PEDV infection in Vero cells even in the absence of type I IFN production.
IKBKB contains an N-terminal kinase domain, a ubiquitin-like domain, an elongated a-helical scaffold/dimerization domain and a NEMO binding domain [
13]. NEMO has also been known as an essential protein for IKBKB activation. NEMO binds to the NEMO binding domain of IKBKB for efficient phosphorylation of IκBα [
3]. To examine the interacting domains of ORF3 and IKBKB, we performed co-immunoprecipitation assays and found that the IKBKB kinase domain alone was not sufficient to bind ORF3. It will be interesting to further investigate if IKBKB kinase activity is important for IKBKB and ORF3 association. In addition, as IKBKB and NEMO interaction could not be displaced by an increasing concentration of ORF3, we thus concluded that the observed interaction between ORF3 and NBD of IKBKB is inadequate to claim the ORF3’s role in competing with NEMO function. To this end, we speculated that the presence of ORF3 might interrupt or disturb IKBKB dimerization, resulting in the perturbation of downstream signaling cascade.
Viral infection is one of the most potent stimuli, which trigger the NF-κB pathway through IKBKB activation. Viruses are capable of both inhibiting and activating NF-κB signal to support their productivity and pathogenesis. Accessory proteins from other coronaviruses, middle east respiratory syndrome coronavirus (MERS-CoV) 4a, 4b, and 8b have been revealed to inhibit MDA5- and TBK1-mediated NF-κB activation, respectively [
25]. Also, severe acute respiratory syndrome coronavirus (SARS-CoV) 8a and 8ab bound IRF3 and accelerated IRF3 degradation through a ubiquitin-proteasome pathway, resulting in reduced IFN-β expression and facilitating virus replication [
26]. Many viral proteins such as HBx protein of hepatitis B virus [
27], nonstructural protein 3 (nsP3) of Venezuelan equine encephalitis virus (VEEV) [
24] and vFLIP protein of Kaposi’s sarcoma-associated herpesvirus (KSHV) [
28] have been reported to manipulate IKBKA or IKBKB and consequentially activate the NF-κB cascade. The aforementioned investigations on coronavirus accessory proteins and other viral proteins point to the potential roles of PEDV ORF3 protein in manipulating both NF-κB and type I IFN signaling. To investigate the effect of ORF3 overexpression on the IKBKB-mediated NF-ҡB promoter activity, cells were treated with TNF-α, a potent NF-ҡB stimulator. Optimized concentrations and durations of TNF-α treatment that triggered IkBα phosphorylation and provided distinguishable proinflammatory cytokine mRNA expression between experimental groups in HEK293T and LLC-PK1 cells were chosen for the study. Consistent with previous finding [
17], our study showed that ORF3 inhibited NF-κB promoter activity [
17] and down-regulated IL-8 and TNF-α mRNA expression. However, when IKBKB and ORF3 were co-expressed, NF-κB promoter activity was increased (
Figure 4A, left panel), whilst IFN-β promoter activity was suppressed (
Figure 6A, left panel). These observations prompt us to speculate that the IKBKB–ORF3 interaction might regulate NF-ҡB and type I IFN signaling pathways via distinct modes of action.
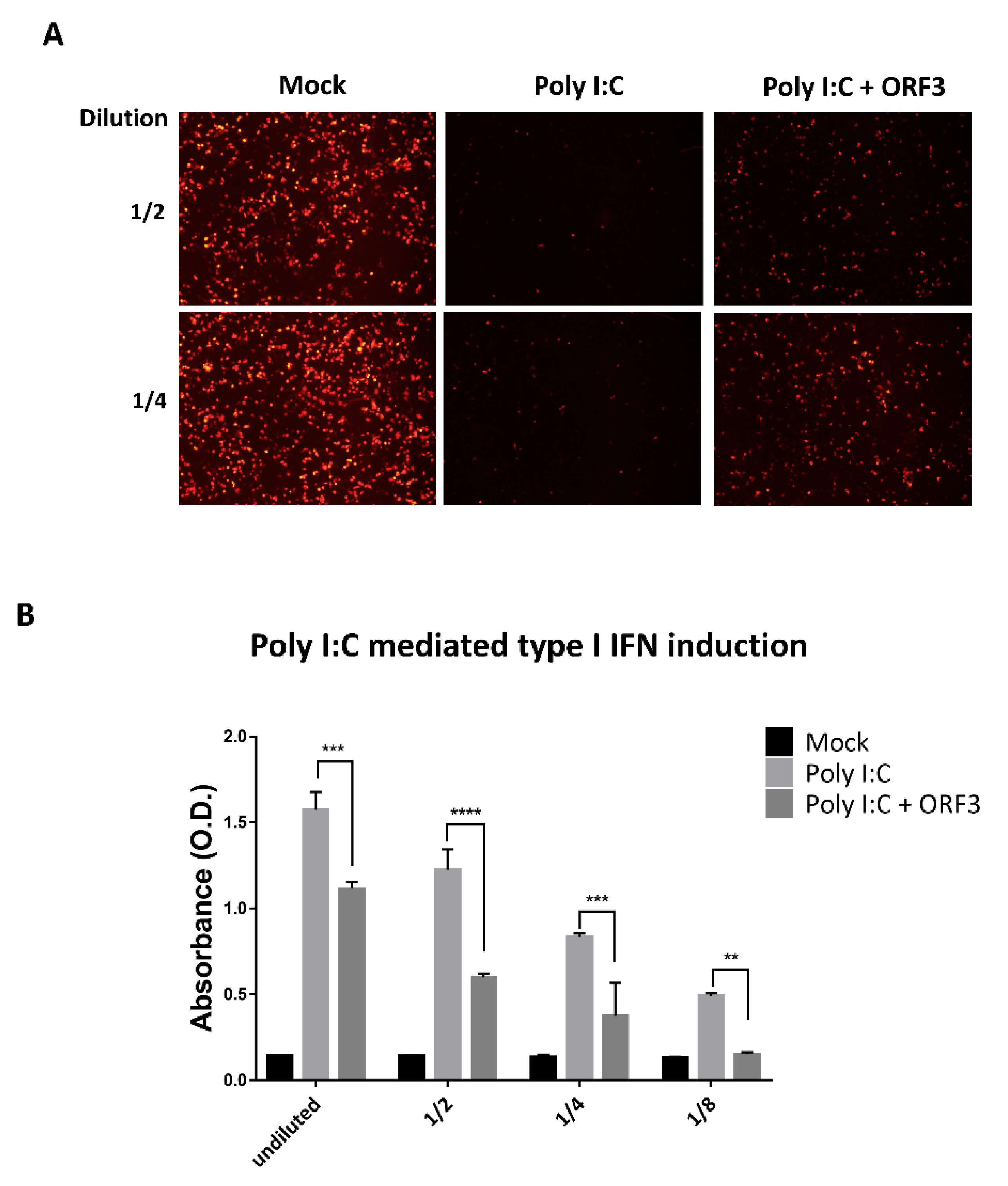
In addition, we found that ORF3 was shown to elevate IKBKB mediated type I IFN induction (
Figure 6A, right panel), but it had a robust negative effect against Poly I:C (
Figure 5B) or RIG-I-mediated type I IFN induction ((
Figure 6B, right panel). Despite the negative impacts ORF3 has on Poly I:C and RIG-I-mediated type I IFN induction, further experiments are still needed to pinpoint which type I IFN signaling intermediate(s) is targeted by ORF3. The discrepancy observed between NF-κB and type I IFN signaling pathways may reflect the importance of a fine balance in the relative amounts of signaling intermediates involved in signal transduction. The interplay between the key signaling proteins might be perturbed or disrupted by ORF3 in the context of PEDV infection. We also speculate that the differential regulation of PEDV ORF3 on both immune signaling pathways is a result of both direct and indirect interaction of ORF3 and IKBKB. Furthermore, it is likely that the ambiguous effects of ORF3 on both NF-κB and type I IFN inductions are due to the fact that IKBKB is able to interact with several other cellular signaling pathways [
13]. Conversely, IKBKB could be one of the many other key transcription factors whose functions are also altered in the presence of ORF3. It is worth mentioning that although the HEK293T cells used in this study serve as a reliable transfection cell culture model, other porcine cell lines which are permissive and biologically relevant to PEDV infection such as IPEC-J2, a porcine jejunal cell line derived from a neonatal pig and its derivative (IPEC-DQ) [
20], could be employed.
In conclusion, we identified an emerging role of PEDV ORF3 as a regulator of IKBKB mediated NF-κB and IFN-β promoter activation, proinflammatory cytokines expression and type I IFN signaling cascade. A schematic representation shown in
Figure 7 summarizes the effects of ORF3 observed in our study on both innate immune signaling pathways. Our present study provides a possible molecular mechanism of ORF3 in manipulating and impeding host immune response and broadens our understanding of ORF3’s role in PEDV pathogenesis. While this study unveiled one of the functions of ORF3 in facilitating PEDV pathogenesis, further investigation is still needed to uncover an as yet unknown involvement of this accessory protein in other aspects of PEDV biology and cellular immune signaling pathways.
4. Materials and Methods
4.1. Cell Lines and Viruses
The African green monkey kidney cell line VeroE6 and human embryonic kidney 293T (HEK293T) cells were cultured in Opti-MEM
® (Gibco
®, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere of 5% CO
2. Porcine cell line LLC-PK1 (ATCC
® CL-101™) was maintained in Medium 199 containing 1.5 g/L to 2.2 g/L sodium bicarbonate supplemented with 3% FBS at 37 °C in a humidified atmosphere of 5% CO
2. PEDV
AV12_ORF3 [
12] was propagated in VeroE6 cells in Opti-MEM
® containing recombinant trypsin (2 µg/mL) (Thermo Scientific, Waltham, MA, USA). The recombinant vesicular stomatitis virus (VSV) expressing the mCherry protein (VSV-mCherry) constructed in our laboratory was previously described [
29].
4.2. Plasmid Construction
pCAGGS plasmids expressing NF-kappa-B essential modulator (NEMO) with hemagglutinin (HA)-tag at the N-terminus (HA-NEMO), an inhibitor of nuclear factor kappa-B kinase subunit beta with Flag-tag at the C-terminus (IKBKB-Flag) [
7] were used as a template for making IKBKB domain deletion, as shown in
Figure 2. All plasmids were subject to direct nucleotide sequencing (First Base, Selangor, Malaysia) and western blot analysis for protein expression.
4.3. Dual-Luciferase Assay
HEK293T cells were plated in 24-well plates at a density of 3 × 105 cells/well and co-transfected with the plasmids expressing firefly luciferase driven under NF-ҡB (pNF-Luc) or IFN-β promoter, IKBKB-Flag, ORF3-Myc and Renilla luciferase (as an internal control) and mock-transfected. At 24 h post transfection (hpt), cells were treated or untreated with TNF-α for 8 hrs, then lysed and subjected for dual-luciferase assay (Promega, Madison, WI, USA) following the manufacturer’s instructions. The data represented relative firefly luciferase activity normalized to Renilla luciferase activity.
4.4. Real-Time PCR Amplification of Proinflammatory Cytokines
Total RNA was isolated from LLC-PK1 cells transfected with plasmids expressing ORF3-Myc or/and IKBKB-Flag by using RNeasy kit (Qiagen, Venlo, Netherlands) and then subjected to RT-qPCR using Luna
® Universal One-Step RT-qPCR Kit (NEB, Ipswich, MA, USA) according to the manufacturer’s protocol. RT-qPCR was performed using the CFX96 Touch™ Real-Time PCR Detection System (BioRad, Hercules, CA, USA). The housekeeping gene β-actin was used as an internal control. Normalized data from each sample relative to zero were compared by the threshold cycle (∆∆
CT) method [
30]. All primers targeting porcine proinflammatory cytokine mRNA included sTNF-α (F: 5′-AACCTCAGATAAGCCCGTCG-3′ and R: 5′-ACCACCAGCTGGTTGTCTTT-3′), sIL−6 (5′-CTGGCAGAAAACAACCTGAACC-3′ and R: 5′-TGATTCTCATCAAGCAGGTCTCC-3′), sIL−8 (F: 5’-CCGTGTCAACATGACTTCCAA–3’ and R: 5’-GCCTCACAGAGAGCTGCAGAA-3’), sIL−1β (F: 5’-ACCTGGACCTTGGTTCTCTG-3’ and R: 5’-CATCTGCCTGATGCTCTTGT-3’) and β-actin (F: 5’-ATCGTGCGTGACATTAAG-3’ and R: 5’-ATTGCCAATGGTGATGAC-3’.
4.5. Confocal Microscopy
The plasmids expressing IKBKB-Flag and ORF3-Myc were co-transfected into VeroE6 and LLC-PK1 cells using Fugene® HD transfection reagent (Promega) following the manufacturer’s instructions. At 24 hpt, cells were fixed with 4% paraformaldehyde in PBS for 20 min at 4 °C. Subsequently, cells were washed three times with PBS and blocked with PBS containing 10% FBS, 1% BSA, and 0.2% TritonX-100 for 1 h. Cells were then incubated for 1 h with rabbit anti-Flag (Abcam, Cambridge, MA, USA) and mouse anti-Myc antibodies (Invitrogen, Carlsbad, CA, USA) diluted in 10% FBS at a dilution of 1:500 and 1:500, respectively. Goat anti-rabbit IgG Alexa Fluor 488 and anti-mouse IgG Alexa Fluor 647 antibodies (both from Abcam) in 10% FBS at a dilution of 1:1000 were used as secondary antibodies and further incubated for 1 h. The glass slips were mounted on slides with ProLongTM Gold Antifade Mountant with DAPI (Invitrogen). The samples were analyzed by FluoviewTM FV1000 confocal microscopy (Olympus, Tokyo, Japan).
4.6. Virus Infection
PEDV
AV12_ORF3 virus [
12] was adsorbed onto VeroE6 cells grown in a 6-well plate. After incubation for 1 h, the cells were washed twice with PBS and maintained in Opti-MEM
® supplemented with recombinant trypsin (2 µg/mL) (Thermo Scientific). Cell supernatants were collected at indicated time points for virus titration (syncytium forming unit (SFU)/mL) as previously described [
12]. Briefly, serial 10-fold dilutions of the virus were inoculated into VeroE6 cells grown in 6-well plates for 24 hpi. Cells were then fixed and subjected to alkaline phosphatase assay detecting system using mouse anti-PEDV N antibodies (Medgene, Brookings, SD, USA) and goat anti-mouse IgG alkaline phosphatase antibodies (Abcam). Numbers of syncytium were counted based on color formation after the addition of 1-Step™ NBT/BCIP Substrate Solution (Thermo Scientific).
4.7. Co-Immunoprecipitation
To confirm physical interaction between ORF3 and IKBKB, HEK293T cells were co-transfected with pCAGGS plasmids expressing ORF3-Myc and IKBKB-Flag or vice versa (ORF3-Flag and IKBKB-Myc for reverse co-immunoprecipitation). At 24 hpt, the cells were washed with cold phosphate buffer saline (PBS) and re-suspended in pre-cooled IP lysis buffer (PierceTM, Thermo Scientific) supplemented with protease inhibitor cocktail (Thermo Scientific). Cell lysates were cleared by centrifugation at 14,000× g for 5 min at 4 °C. Cleared lysates were then incubated with anti-Myc agarose (PierceTM, Thermo scientific) with gentle rocking overnight at 4 °C. Immunoprecipitates were washed with TBST buffer (25 mM Tris-HCl, 0.15 M NaCl, 0.05% tween 20, pH 7.2) and eluted in sodium dodecyl sulfate (SDS) sample loading buffer. The samples were subjected to SDS-PAGE and western blot.
For ORF3 and NEMO competition assay, each of plasmids expressing ORF3-Flag, IKBKB-Myc and HA-NEMO were transfected into HEK293T cells. At 24 hpt, cells were collected and lysed with pre-cooled IP lysis buffer (PierceTM, Thermo Scientific). An equal amount of lysate containing IKBKB-Myc and HA-NEMO and varied amounts of lysate with ORF3-Flag was combined and subjected to immunoprecipitation using anti-Myc bead as described above. Immunoprecipitates were eluted in sample buffer followed by SDS-PAGE and western blot.
4.8. Western Blot Analysis
Cells were collected and re-suspended in the PierceTM mammalian cell lysis buffer (PierceTM, Thermo Scientific). Protein samples were mixed with SDS sample loading buffer, loaded onto 10% or 12% polyacrylamide gel, and transferred to nitrocellulose membranes. Membranes were incubated with rabbit anti-Myc (Abcam) antibody, rabbit anti-Flag (Abcam) antibody and rabbit anti-HA antibody. Rabbit anti-IKBKB antibody (Abcam) was used to detect endogenous IKBKB. Mouse anti-β-actin (Cell signaling) was used to detect β-actin for internal controls. Goat anti-rabbit IgG-HRP (KPL, MA, USA) and anti-mouse IgG-HRP antibodies were used as secondary antibodies for chemiluminescence detection by ChemiDoc™ XRS+ imager (BioRad-).
4.9. IFN Bioassay
Levels of IFN secreted by transfected HEK293T cells were determined as previously described [
31,
32], with some modification. HEK293T cells seeded in 6-well-plates one day before transfection to reach 80%–90% confluence. These cells were either mock-treated or transfected with ORF3, IKBKB, a combination of ORF3 and IKBKB expressing plasmids, and poly I:C by Fugene
® Transfection Reagent (Promega). Following transfection, cells were incubated in Opti-MEM
® containing 10% FBS, and supernatants were harvested 24 h post infection (hpi). PK-15 cells were seeded in 96-well plates one day before supernatant harvest. These PK-15 cells were then incubated with the harvested supernatants for 24 hours. The preincubated PK-15 cells were then infected with VSV-mCherry (MOI = 1). Cells expressing mCherry were visualized by fluorescence microscopy at 16 hpi. The presence of IFN in the supernatants was estimated based on their abilities to induce an antiviral state in PK-15 cells and inhibit VSV–mCherry replication, compared with known amounts of recombinant porcine IFN.
4.10. ISRE/SEAP Activity Quantification
Type I IFNs in supernatants harvested from transfected HEK cells were quantified by HEK-BlueTM IFN-α/β cells (Invivogen, San Diego, CA, USA) containing interferon-sensitive response element (ISRE) promoter. The presence of type I IFNs in supernatant induces ISRE promoter activation, which could be evaluated by using the substrate Qanti-blue (Invivogen) according to the manufacturer’s recommendations to measure secreted embryonic alkaline phosphatase (SEAP) activity. As a positive control, HEK-BlueTM IFN-α/β cells were treated for 24 hours with supernatant of Poly I:C transfected HEK cells.
4.11. Statistical Analysis
All data were expressed as means ± standard error of means (SEM). The differences in mean values of between groups were analyzed by two-way ANOVA. p values of <0.05 were considered statistically significant. GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) was used for statistical analyses.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






