Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia

 ,
,  ,
, 
 and
and
Abstract
:1. Introduction
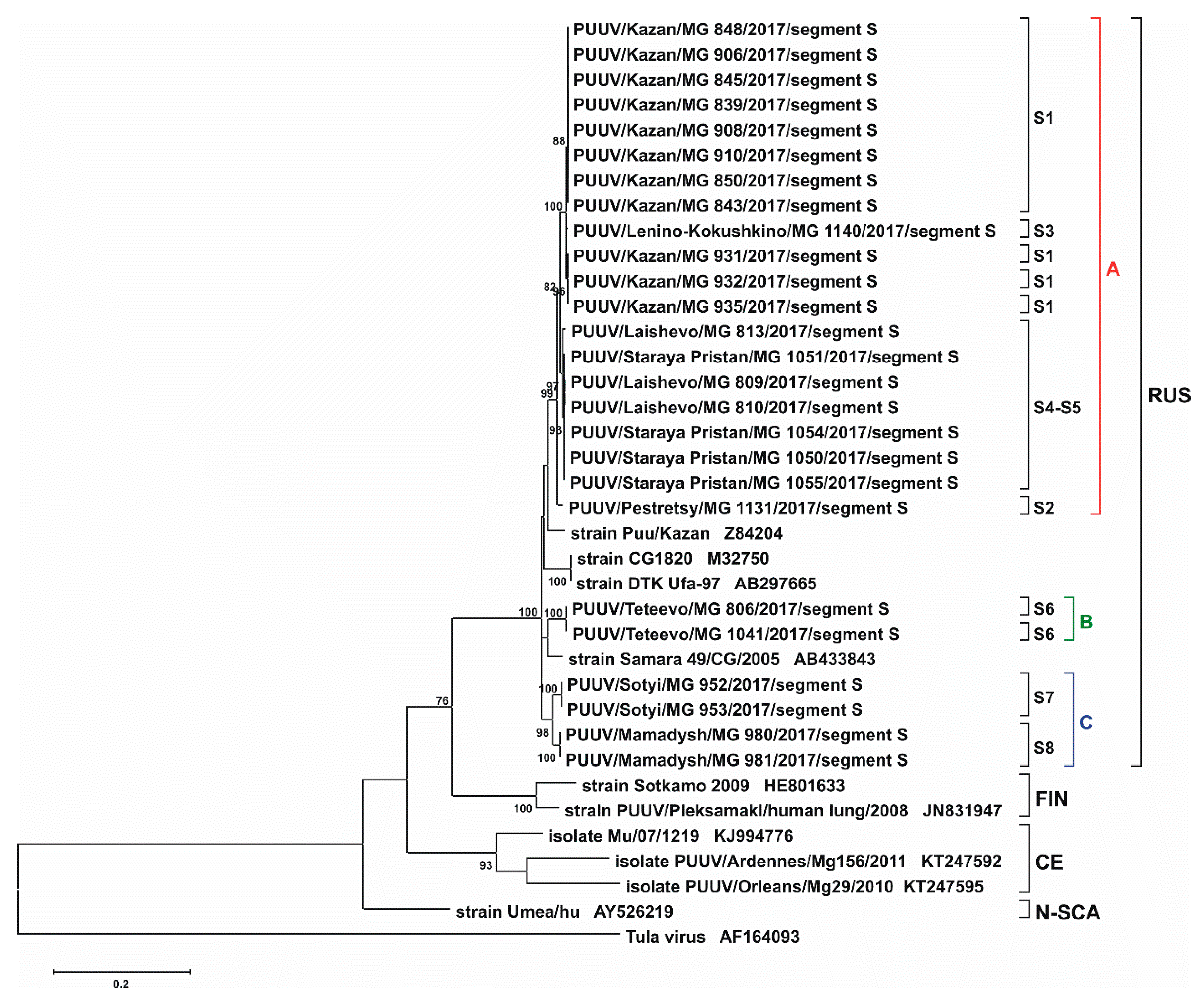
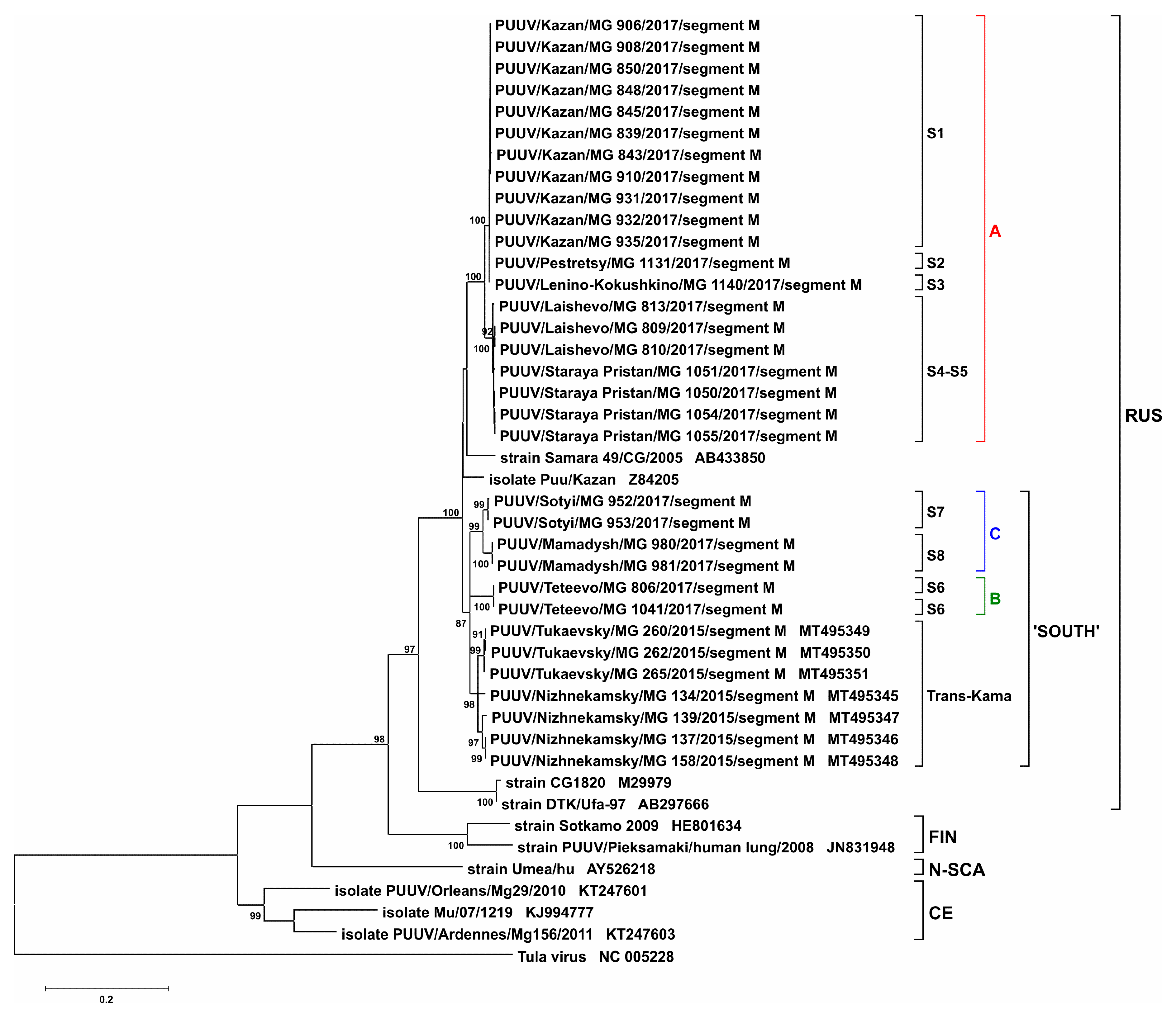
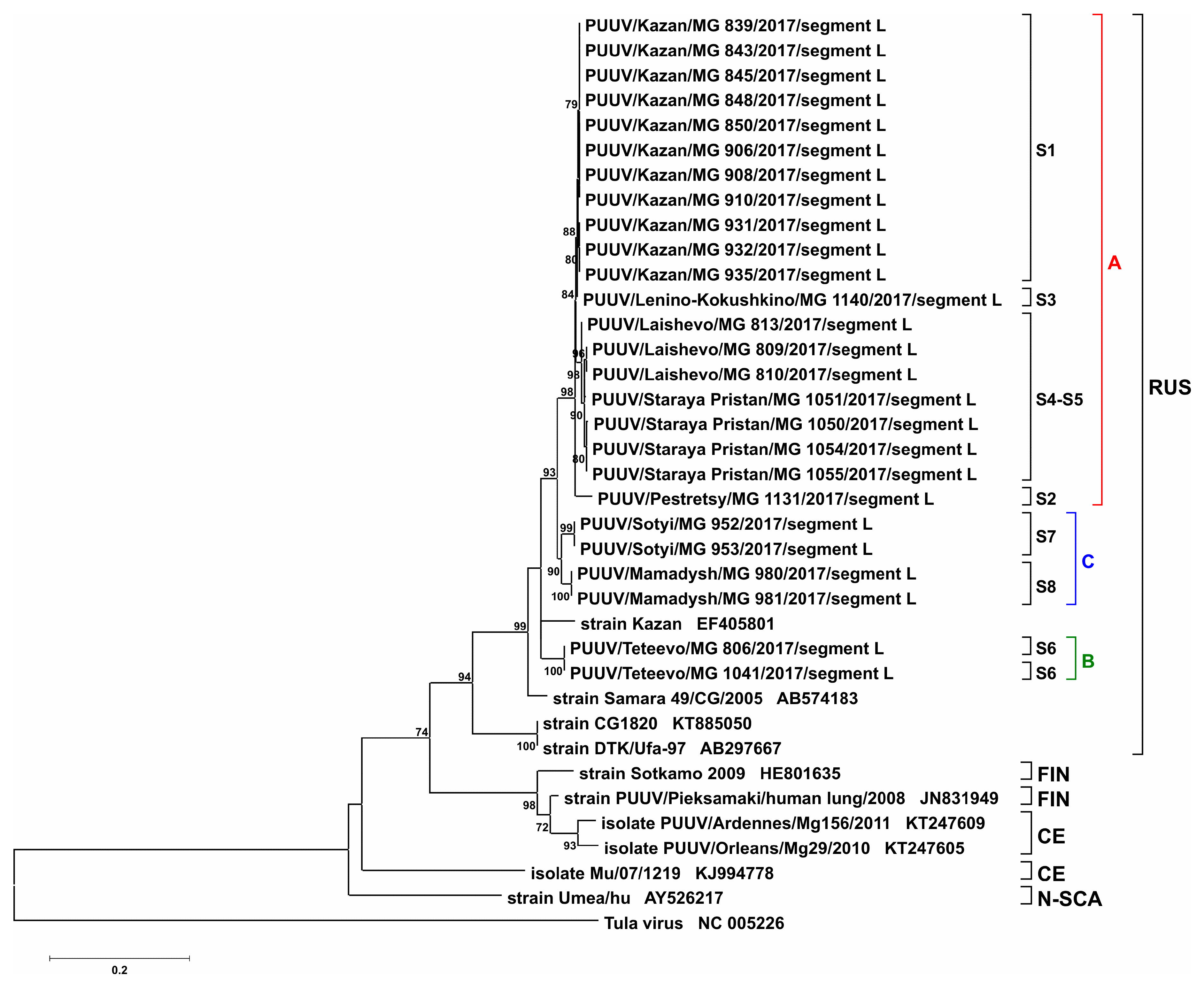
2. Results and Discussion
3. Materials and Methods
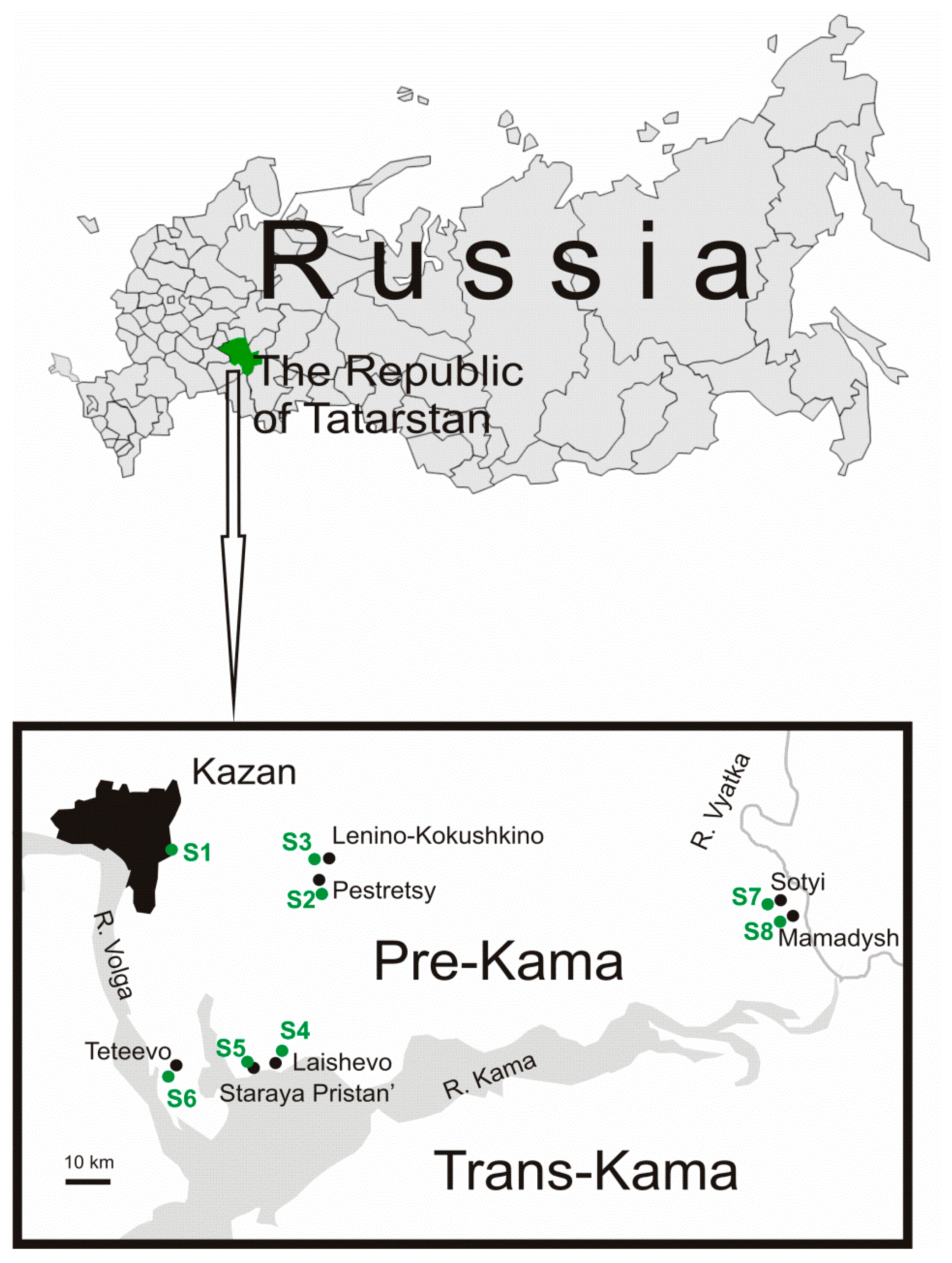
3.1. Bank Vole Trapping and Abundance
3.2. RNA extraction, cDNA synthesis and Polymerase Chain Reaction (PCR)
3.3. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kruger, D.H.; Figueiredo, L.T.M.; Song, J.-W.; Klempa, B. Hantaviruses—globally emerging pathogens. J. Clin. Virol. 2015, 64, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Mustonen, J.; Lundkvist, Å.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Jiang, H.; Zheng, X.; Wang, L.; Du, H.; Wang, P.; Bai, X. Hantavirus infection: A global zoonotic challenge. Virol. Sin. 2017, 32, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Vapalahti, O.; Vaheri, A. Hantaviruses: Genome structure, expression and evolution. J. Gen. Virol. 1996, 77, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Milholland, M.T.; Castro-Arellano, I.; Suzán, G.; Garcia-Peña, G.E.; Lee, T.E.; Rohde, R.E.; Aguirre, A.A.; Mills, J.N. Global diversity and distribution of hantaviruses and their hosts. Ecohealth 2018, 15, 163–208. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, J.; Mäkelä, S.; Outinen, T.; Laine, O.; Jylhävä, J.; Arstila, P.T.; Hurme, M.; Vaheri, A. The pathogenesis of nephropathia epidemica: New knowledge and unanswered questions. Antiviral Res. 2013, 100, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariwa, H.; Tkachenko, E.A.; Morozov, V.G.; Seto, T.; Tanikawa, Y.; Kolominov, S.I.; Belov, S.N.; Nakamura, I.; Hashimoto, N.; Balakiev, A.E.; et al. Epidemiological study of hantavirus infection in the Samara Region of European Russia. J. Vet. Med. Sci. 2009, 71, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Ivanova, A.V.; Popov, N.V.; Pakskina, N.D.; Kuznetsov, A.A.; Matrosov, A.N.; Shylov, M.M.; Mochalkin, P.A.; Korneev, M.G.; Toporkov, V.P. Epidemiological activity of natural foci of hemorrhagic fever with renal syndrome on the territory of the Russian Federation in 2013–2017 and forecast for 2018. Probl. Espec. Danger. Infect. 2018. [Google Scholar] [CrossRef] [Green Version]
- Korneev, M.G.; Chekashov, V.N.; Ivanova, A.V.; Matrosov, A.N.; Kuznetsov, A.A.; Shylov, M.M. Review of the Number of Carriers and Vectors of Zoonoses, Epizootic and Epidemiological Situation in the Volga Federal District in Second Half 2019 and Forecast for 2020. (In Russian). Available online: http://www.microbe.ru/files/PFO_revII_2019_prognI_2020.pdf (accessed on 3 July 2020).
- Stupishyn, A.V.; Babanov, Y.V.; Guseva, A.A. Physico-geographical zoning of the Middle Volga. Kazan; Kazan University Publishing: Kazan, Russia, 1964; p. 197. (In Russian) [Google Scholar]
- Boiko, V.A.; Trifonov, A.V.; Reshetnikova, I.D.; Agafonova, E.V.; Potapov, V.S.; Gubeidullina, A.K.h.; Kryuchkov, R.A.; Shamsutdinov, A.F. Monitoring of Dominant Natural Focal Infections and the Experience of Assessing the Potential Risk of Infection of the Population in the Landscape Subzones of Tatarstan: A Collective Monograph; “Medicine”: Kazan, Russia, 2017; p. 83. (In Russian) [Google Scholar]
- Kabwe, E.; Davidyuk, N.; Morzunov, S.; Shakirova, V.; Anokhin, V.; Isaeva, G.; Ismagilova, R.; Khaiboullina, S.; Rizvanov, A. Genetic characterization of small (s)-segment genome Puumala virus strain Kazan. BioNanoScience 2017, 7, 316–319. [Google Scholar] [CrossRef]
- Kabwe, E.; Davidyuk, Y.N.; Morzunov, S.P. Genome variations of Puumala virus strains circulating in Nizhnekamsky and Tukaevsky districts of the Republic of Tatarstan. Uchenye Zapiski Kazanskogo Universiteta. Seriya Estestvennye Nauki 2018, 160, 373–385. [Google Scholar]
- Davidyuk, Y.; Kabwe, E.; Shakirova, V.; Martynova, E.V.; Ismagilova, R.; Khaertynova, I.; Khaiboullina, S.; Rizvanov, A.; Morzunov, S.P. Characterization of the Puumala orthohantavirus strains in the northwestern region of the Republic of Tatarstan in relation to the clinical manifestations in hemorrhagic fever with renal syndrome patients. Front. Pharmacol. 2019, 10, 970. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Kim, W.-k.; No, J.S.; Lee, S.-H.; Lee, S.-Y.; Kim, J.H.; Kho, J.H.; Lee, D.; Song, D.H.; Gu, S.H. Genetic diversity and reassortment of Hantaan virus tripartite RNA genomes in nature, the Republic of Korea. PLoS Negl. Trop. Dis. 2016, 10, e0004650. [Google Scholar] [CrossRef] [PubMed]
- Rizvanov, A.A.; Khaiboullina, S.F.; Jeor, S.S. Development of reassortant viruses between pathogenic hantavirus strains. Virology 2004, 327, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Szabó, R.; Radosa, L.; Ličková, M.; Sláviková, M.; Heroldová, M.; Stanko, M.; Pejčoch, M.; Osterberg, A.; Laenen, L.; Schex, S. Phylogenetic analysis of Puumala virus strains from Central Europe highlights the need for a full-genome perspective on hantavirus evolution. Virus Genes 2017, 53, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Pavlinov, I.Y.; Krusko, S.V.; Warsawsky, A.A. Terrestrial mammals of Russia: A reference guide. Sci. Press. Moscow KMK 2002, 1, 298. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Trapping Site | Number of Trapped Bank Voles | Number of RT-PCR Positive | No. of Sequences Used for Analysis a | ||
|---|---|---|---|---|---|---|
| Segment S (nt 240–1296) | Segment M (nt 1499–2512) | Segment L (nt 958–1622) | ||||
| Kazan | S1 | 46 | 12 | 11 | 11 | 11 |
| Pestretsy | S2 | 4 | 1 | 1 | 1 | 1 |
| Lenino-Kokushkino | S3 | 3 | 1 | 1 | 1 | 1 |
| Laishevo | S4 | 6 | 4 | 3 | 3 | 3 |
| Staraya Pristan’ | S5 | 15 | 4 | 4 | 4 | 4 |
| Teteevo | S6 | 29 | 2 | 2 | 2 | 2 |
| Sotyi | S7 | 12 | 3 | 2 | 2 | 2 |
| Mamadysh | S8 | 4 | 2 | 2 | 2 | 2 |
| In total | 119 | 29 | 26 | 26 | 26 | |
| Cluster | A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Site | Direction | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | |
| A | S1 | nt → | 0.0–0.6 | 2.3–2.4 | 0.6 | 1.9–2.1 | 1.8–2.0 | 6.0–6.1 | 6.0–6.2 | 5.5–5.8 |
| aa ↓ | 0.0 | |||||||||
| S2 | 0.0 | 2.4 | 1.7–2.0 | 1.8–1.9 | 5.5–5.6 | 5.9 | 5.1–5.2 | |||
| 0.6 | 0.0 | |||||||||
| S3 | 0.0 | 2.0–2.1 | 1.9–2.0 | 6.1 | 6.2 | 5.7–5.8 | ||||
| 0.0 | 0.6 | 0.0 | ||||||||
| S4 | 0.0–0.8 | 0.1–0.8 | 5.4–5.8 | 5.6–5.9 | 5.1–5.5 | |||||
| 0.6 | 0.0 | 0.6 | 0.0 | |||||||
| S5 | 0.0–0.2 | 5.2–5.3 | 5.4–5.5 | 4.9–5.1 | ||||||
| 0.6 | 0.0 | 0.6 | 0.0 | 0.0 | ||||||
| B | S6 | 0.1 | 6.0–6.1 | 5.9–6.0 | ||||||
| 0.6 | 0.6 | 0.6 | 0.6 | 0.6 | 0.0 | |||||
| C | S7 | 0.0 | 2.2–2.3 | |||||||
| 0.6 | 0.0 | 0.6 | 0.0 | 0.0 | 0.6 | 0.0 | ||||
| S8 | 0.1 | |||||||||
| 0.6 | 0.0 | 0.6 | 0.0 | 0.0 | 0.6 | 0.0 | 0.0 | |||
| Trapping site | Genetic lineage | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RUS | FIN | CE | N-SCA | |||||||
| Name of Strain | ||||||||||
| Samara_49/CG/2005 | Puu/Kazan | CG1820 | DTK/Ufa-97 | Sotkamo 2009 | PUUV/Pieksamaki/human_lung/2008 | Mu/07/1219 | PUUV/Ardennes/Mg156/2011 | PUUV/Orleans/Mg29/2010 | Umea/hu | |
| Segment S | ||||||||||
| S1 | 6.4–6.5 | 5.1–5.3 | 6.3–6.6 | 6.2–6.5 | 17.6–17.7 | 16.4–16.5 | 19.7–19.9 | 19.2–19.6 | 21.2–21.3 | 18.4–18.7 |
| S2 | 6.1 | 4.5 | 5,8 | 5.7 | 17.3 | 16.4 | 18.7 | 19.4 | 21.3 | 18.7 |
| S3 | 6.5 | 5.3 | 6.6 | 6.5 | 18.0 | 16.7 | 19.8 | 19.7 | 21.3 | 18.7 |
| S4 | 5.8–6.1 | 4.6–4.7 | 6.4–6.7 | 6.3–6.6 | 17.3–18.1 | 16.6–16.7 | 19.1–19.4 | 19.3–19.6 | 21.0–21.1 | 18.7–19.0 |
| S5 | 5.9–6.1 | 4.4–4.5 | 6.2–6.4 | 6.1–6.3 | 17.8–18.1 | 16.5–16.7 | 19.3–19.4 | 19.2–19.3 | 20.9–21.0 | 18.7–19.0 |
| S6 | 4.8–4.9 | 6.1–6.2 | 6.6–6.7 | 6.5–6.6 | 18.2 | 15.2–16.3 | 18.7 | 20.3–20.5 | 21.8–21.9 | 18.4 |
| S7 | 4.8 | 5.4 | 6.6 | 6.5 | 17.1 | 15.4 | 19.5 | 19.2 | 21.3 | 19.5 |
| S8 | 4.7–4.8 | 4.8–4.9 | 6.6–6.7 | 6.5–6.6 | 16.8 | 15.4 | 18.4 | 19.2 | 21.2 | 19.7–19.9 |
| Segment M | ||||||||||
| S1 | 6.6–6.7 | 6.3–6.6 | 16.0–16.1 | 15.3–15.4 | 18.0–18.4 | 18.9–19.4 | 23.1–23.4 | 21.6–22.0 | 25.1–25.2 | 22.0–22.2 |
| S2 | 6.6 | 6.3 | 16.0 | 15.3 | 18.0 | 18.9 | 23.0 | 21.6 | 25.1 | 22.0 |
| S3 | 6.6 | 6.3 | 15.7 | 15.0 | 18.0 | 18.9 | 23.1 | 21.6 | 24.8 | 22.0 |
| S4 | 7.2–7.6 | 6.5–6.6 | 16.2–16.4 | 15.5–15.7 | 18.5–18.8 | 19.6–19.8 | 23.5–23.7 | 21.8–22.5 | 24.1–24.2 | 22.9–23.2 |
| S5 | 7.6–7.7 | 6.7–6.8 | 16.2–16.6 | 15.5–15.9 | 18.4–18.8 | 19.6–20.0 | 23.8–24.1 | 22.2–22.3 | 24.4–24.8 | 23.0–23.2 |
| S6 | 8.6–8.7 | 7.0–7.1 | 17.3 | 16.5 | 18.7 | 18.8–18.9 | 24.6–24.7 | 21.5–21.6 | 23.5–23.6 | 22.1–22.3 |
| S7 | 7.3–7.4 | 6.5 | 14.6 | 13.9 | 17.7 | 18.1 | 24.3–24.4 | 21.2–21.3 | 23.6–23.8 | 21.0–21.3 |
| S8 | 7.8 | 6.9 | 14.9 | 14.2 | 18.1 | 17.7 | 24.4 | 21.3 | 24.4 | 22.1 |
| Segment L | ||||||||||
| S1 | 7.4 | 7.2–7.5 | 14.5–14.9 | 14.5–14.9 | 17.6–18.0 | 18.4–18.8 | 20.7–21.1 | 21.0–21.5 | 20.2–20.6 | 22.7–23.2 |
| S2 | 7.7 | 8.9 | 15.3 | 15.3 | 19.4 | 19.6 | 20.0 | 22.3 | 20.8 | 23.3 |
| S3 | 7.4 | 7.5 | 14.9 | 14.9 | 18.0 | 18.4 | 21.1 | 21.0 | 20.2 | 23.2 |
| S4 | 7.7–8.3 | 8.1–8.9 | 14.9–15.9 | 14.9–15.9 | 18.6–18.8 | 18.6–18.8 | 21.3 | 21.7–21.9 | 20.4–20.6 | 24.0–24.5 |
| S5 | 8.1–8.6 | 8.4–8.9 | 15.3–15.9 | 15.3–15.9 | 18.4–18.64 | 18.4–18.6 | 21.3–21.9 | 21.5–21.7 | 20.2–20.6 | 24.3–24.7 |
| S6 | 6.7 | 6.7 | 14.9 | 14.9 | 19.2 | 18.6 | 21.0 | 20.8 | 19.1 | 23.1 |
| S7 | 7.6 | 7.4 | 14.5 | 14.5 | 19.4 | 18.8 | 22.1 | 21.0 | 20.2 | 22.9 |
| S8 | 6.7 | 7.7 | 14.3 | 14.3 | 18.4 | 17.6 | 21.9 | 20.4 | 19.5 | 23.4 |
| Cluster | A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Site | Direction | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | |
| A | S1 | nt → | 0.0–0.3 | 0.2–0.5 | 0.2–0.5 | 2.3–2.7 | 2.3–2.7 | 8.1–8.4 | 6.7–7.0 | 7.0–7.3 |
| aa ↓ | 0.0 | |||||||||
| S2 | 0.0 | 0.2 | 2.3–2.4 | 2.3–2.4 | 8.1–8.3 | 6.8–6.9 | 7.1 | |||
| 0.3 | 0.0 | |||||||||
| S3 | 0.0 | 2.3–2.4 | 2.3–2.4 | 8.1–8.3 | 6.8–6.9 | 7.1 | ||||
| 0.3 | 0.0 | 0.0 | ||||||||
| S4 | 0.0–0.5 | 0.3–0.5 | 8.0–8.5 | 6.6–7.0 | 7.7–7.8 | |||||
| 0.3 | 0.0 | 0.0 | 0.0 | |||||||
| S5 | 0.0–0.3 | 8.3–8.5 | 6.8–7.0 | 7.7–7.8 | ||||||
| 0.3 | 0.0 | 0.0 | 0.0 | 0.0 | ||||||
| B | S6 | 0.1 | 5.8–6.1 | 6.2–6.4 | ||||||
| 0.9–1.2 | 0.6–0.9 | 0.6–0.9 | 0.6–0.9 | 0.6–0.9 | 0.3 | |||||
| C | S7 | 0.2 | 2.1–2.3 | |||||||
| 0.6 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3–0.6 | 0.0 | ||||
| S8 | 0.0 | |||||||||
| 0.9 | 0.6 | 0.6 | 0.6 | 0.6 | 0.6–0.9 | 0.3 | 0.0 | |||
| Trapping site | Genetic lineage | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RUS | FIN | CE | N-SCA | |||||||
| Name of Strain | ||||||||||
| Samara_49/CG/2005 | Puu/Kazan | CG1820 | DTK/Ufa-97 | Sotkamo 2009 | PUUV/Pieksamaki/human_lung/2008 | Mu/07/ 1219 | PUUV/Ardennes/Mg156/2011 | PUUV/Orleans/Mg29/2010 | Umea/hu | |
| Segment S | ||||||||||
| S1 | 0.6 | 1.1 | 1.4 | 1.1 | 3.5 | 3.2 | 2.6 | 3.5 | 4.4 | 3.2 |
| S2 | 0.6 | 0.6 | 0.9 | 0.6 | 2.9 | 2.6 | 2.0 | 2.9 | 3.6 | 2.6 |
| S3 | 0.6 | 1.1 | 1.4 | 1.1 | 3.5 | 3.2 | 2.6 | 3.5 | 4.4 | 3.2 |
| S4 | 0.6 | 0.6 | 0.9 | 0.6 | 2.9 | 2.6 | 2.0 | 2.9 | 3.8 | 2.6 |
| S5 | 0.6 | 0.6 | 0.9 | 0.6 | 2.9 | 2.6 | 2.0 | 2.9 | 3.8 | 2.6 |
| S6 | 0.0 | 1.1 | 0.9 | 0.6 | 3.5 | 3.2 | 2.3 | 2.9 | 4.1 | 3.2 |
| S7 | 0.6 | 0.6 | 0.9 | 0.6 | 2.9 | 2.6 | 2.0 | 2.9 | 3.8 | 2.6 |
| S8 | 0.6 | 0.6 | 0.9 | 0.6 | 2.9 | 2.6 | 2.0 | 2.9 | 3.8 | 2.6 |
| Segment M | ||||||||||
| S1 | 1.2 | 0.9 | 3.6 | 2.1 | 3.6 | 3.6 | 9.1 | 7.5 | 8.1 | 9.1 |
| S2 | 0.9 | 0.6 | 3.3 | 1.8 | 3.3 | 3.3 | 8.8 | 7.1 | 7.8 | 8.8 |
| S3 | 0.9 | 0.6 | 3.3 | 1.8 | 3.3 | 3.3 | 8.8 | 7.1 | 7.8 | 8.8 |
| S4 | 0.9 | 0.6 | 3.3 | 1.8 | 3.3 | 3.3 | 8.8 | 7.1 | 7.8 | 8.8 |
| S5 | 0.9 | 0.6 | 3.3 | 1.8 | 3.3 | 3.3 | 8.8 | 7.1 | 7.8 | 8.8 |
| S6 | 0.9–1.2 | 0.6–0.9 | 3.3–3.6 | 1.8–2.1 | 3.3–3.6 | 3.3–3.6 | 9.1–9.5 | 7.5–7.8 | 8.1–8.5 | 8.8–9.1 |
| S7 | 0.6 | 0.3 | 3.0 | 1.5 | 3.0 | 3.0 | 8.8 | 7.1 | 7.8 | 8.5 |
| S8 | 0.9 | 0.6 | 3.3 | 1.8 | 3.3 | 3.3 | 9.1 | 7.5 | 8.1 | 8.8 |
| Segment L | ||||||||||
| S1 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| S2 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| S3 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| S4 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| S5 | 0.9–1.4 | 0.5–0.9 | 1.4–1.8 | 1.4–1.8 | 5.6–6.1 | 6.1–6.6 | 9.2–9.7 | 7.6–8.1 | 6.6–7.1 | 10.7–11.2 |
| S6 | 1.4 | 0.9 | 1.8 | 1.8 | 6.6 | 7.1 | 9.7 | 8.6 | 7.6 | 11.2 |
| S7 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| S8 | 0.9 | 0.5 | 1.4 | 1.4 | 6.1 | 6.6 | 9.2 | 8.1 | 7.1 | 10.7 |
| Cluster | A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Site | Direction | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | |
| A | S1 | nt → | 0.0–0.3 | 2.9 | 0.3 | 1.2–2.0 | 1.8–2.0 | 6.5 | 4.9–5.2 | 4.4 |
| aa ↓ | 0.0 | |||||||||
| S2 | 0.0 | 2.6 | 2.9–3.7 | 3.6–3.7 | 8.0 | 7.0 | 5.5 | |||
| 0.0 | 0.0 | |||||||||
| S3 | 0.0 | 0.9–1.7 | 1.5–1.7 | 6.5 | 4.9 | 4.0 | ||||
| 0.0 | 0.0 | 0.0 | ||||||||
| S4 | 0.0–0.8 | 0.5–0.9 | 7.2–8.1 | 5.5–6.4 | 4.7–5.5 | |||||
| 0.0 | 0.0 | 0.0 | 0.0 | |||||||
| S5 | 0.0–0.6 | 7.5–7.9 | 5.9–6.4 | 5.0–5.5 | ||||||
| 0.0–0.5 | 0.0–0.5 | 0.0–0.5 | 0.0–0.5 | 0.0–0.5 | ||||||
| B | S6 | 0.0 | 6.7 | 6.7 | ||||||
| 0.5 | 0.5 | 0.5 | 0.5 | 0.5–0.9 | 0.0 | |||||
| C | S7 | 0.0 | 3.1 | |||||||
| 0.0 | 0.0 | 0.0 | 0.0–0.5 | 0.0–0.5 | 0.5 | 0.0 | ||||
| S8 | 0.0 | |||||||||
| 0.0 | 0.0 | 0.0 | 0.0–0.5 | 0.0–0.5 | 0.5 | 0.0 | 0.0 | |||
| Cluster | Site | Strain | Segment a | ||
|---|---|---|---|---|---|
| partial S (nt 240–1296) | partial M (nt 1499–2512) | partial L (nt 958–1622) | |||
| A | S1 | 839 | A1 | A1 | A1 |
| 843 | A1 | A1 | A1 | ||
| 845 | A1 | A1 | A1 | ||
| 848 | A1 | A1 | A1 | ||
| 850 | A1 | A1 | A1 | ||
| 906 | A1 | A1 | A1 | ||
| 908 | A1 | A1 | A1 | ||
| 910 | A1 | A1 | A1 | ||
| 931 | A1 | A1 | A1 | ||
| 932 | A1 | A1 | A1 | ||
| 935 | A1 | A1 | A1 | ||
| S2 | 1131 | A2 | A1 | A2 | |
| S3 | 1140 | A1 | A1 | A1 | |
| S4 | 809 | A3 | A3 | A3 | |
| 810 | A3 | A3 | A3 | ||
| 813 | A3 | A3 | A3 | ||
| S5 | 1050 | A3 | A3 | A3 | |
| 1051 | A3 | A3 | A3 | ||
| 1054 | A3 | A3 | A3 | ||
| 1055 | A3 | A3 | A3 | ||
| B | S6 | 806 | B | B | B |
| 1041 | B | B | B | ||
| C | S7 | 952 | C1 | C1 | C1 |
| 953 | C1 | C1 | C1 | ||
| S8 | 980 | C2 | C2 | C2 | |
| 981 | C2 | C2 | C2 | ||
| Genome Segment | Primer Name | Sequence (5′→3′) | Position a (nt) | Direction | Reference |
|---|---|---|---|---|---|
| S | PUUV-S-F1 | tagtagtagactccttgaaaagc | 1–23 | Forward | This paper |
| PUUV-S-F41 | agctactacgagaacaactgg | 21–41 | Forward | This paper | |
| PuuV-for | ctgcaagccaggcaacaaacagtgtcagca | 172–201 | Forward | [9] | |
| 4S-F3 | gcactggaggataaactcgc | 199–218 | Forward | This paper | |
| PUUV-S-F704 | aacatcatgagtccagtaatggg | 682–704 | Forward | This paper | |
| 69S-F3 | ttatggcatctaaaactgtgg | 1079–1099 | Forward | This paper | |
| PUUV-S-R1496 | gtataattccagttaacccctg | 1496–1517 | Reverse | This paper | |
| PUUV-S-R1183 | gtacagtaggattatcctctgatc | 1183–1206 | Reverse | This paper | |
| PuuV-Rev | gtctgccacatgatttttgtcaagcacatc | 865–894 | Reverse | [9] | |
| 5S-B3 | ggccagtctttaagcaagaaag | 719–740 | Reverse | This paper | |
| R358 PUUVS | catttacatcaaggacatttcc | 337–358 | Reverse | This paper | |
| M | F1452 PUUVM | tctttaatcccaggagttgc | 1451–1470 | Reverse | [16] |
| R2582 PUUVM | aaattgtccctattaaacacac | 2561–2582 | Reverse | [16] | |
| L | F925-PUUL | agcttccaagcaccatatttaccatc | 925–950 | Forward | This paper |
| R1663-PUUL | gattatctgcatcaataagacctagt | 1638–1663 | Reverse | This paper |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidyuk, Y.; Shamsutdinov, A.; Kabwe, E.; Ismagilova, R.; Martynova, E.; Belyaev, A.; Shuralev, E.; Trifonov, V.; Savitskaya, T.; Isaeva, G.; et al. Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens 2020, 9, 540. https://doi.org/10.3390/pathogens9070540
Davidyuk Y, Shamsutdinov A, Kabwe E, Ismagilova R, Martynova E, Belyaev A, Shuralev E, Trifonov V, Savitskaya T, Isaeva G, et al. Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens. 2020; 9(7):540. https://doi.org/10.3390/pathogens9070540
Chicago/Turabian StyleDavidyuk, Yuriy, Anton Shamsutdinov, Emmanuel Kabwe, Ruzilya Ismagilova, Ekaterina Martynova, Alexander Belyaev, Eduard Shuralev, Vladimir Trifonov, Tatiana Savitskaya, Guzel Isaeva, and et al. 2020. "Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia" Pathogens 9, no. 7: 540. https://doi.org/10.3390/pathogens9070540
APA StyleDavidyuk, Y., Shamsutdinov, A., Kabwe, E., Ismagilova, R., Martynova, E., Belyaev, A., Shuralev, E., Trifonov, V., Savitskaya, T., Isaeva, G., Khaiboullina, S., Rizvanov, A., & Morzunov, S. (2020). Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens, 9(7), 540. https://doi.org/10.3390/pathogens9070540