SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings



Abstract
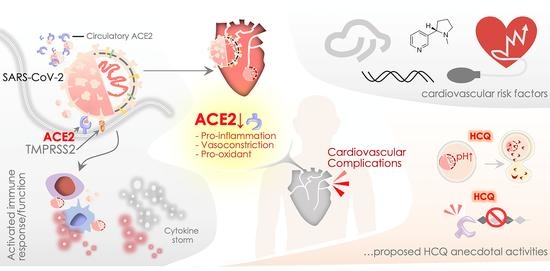
:
1. Introduction
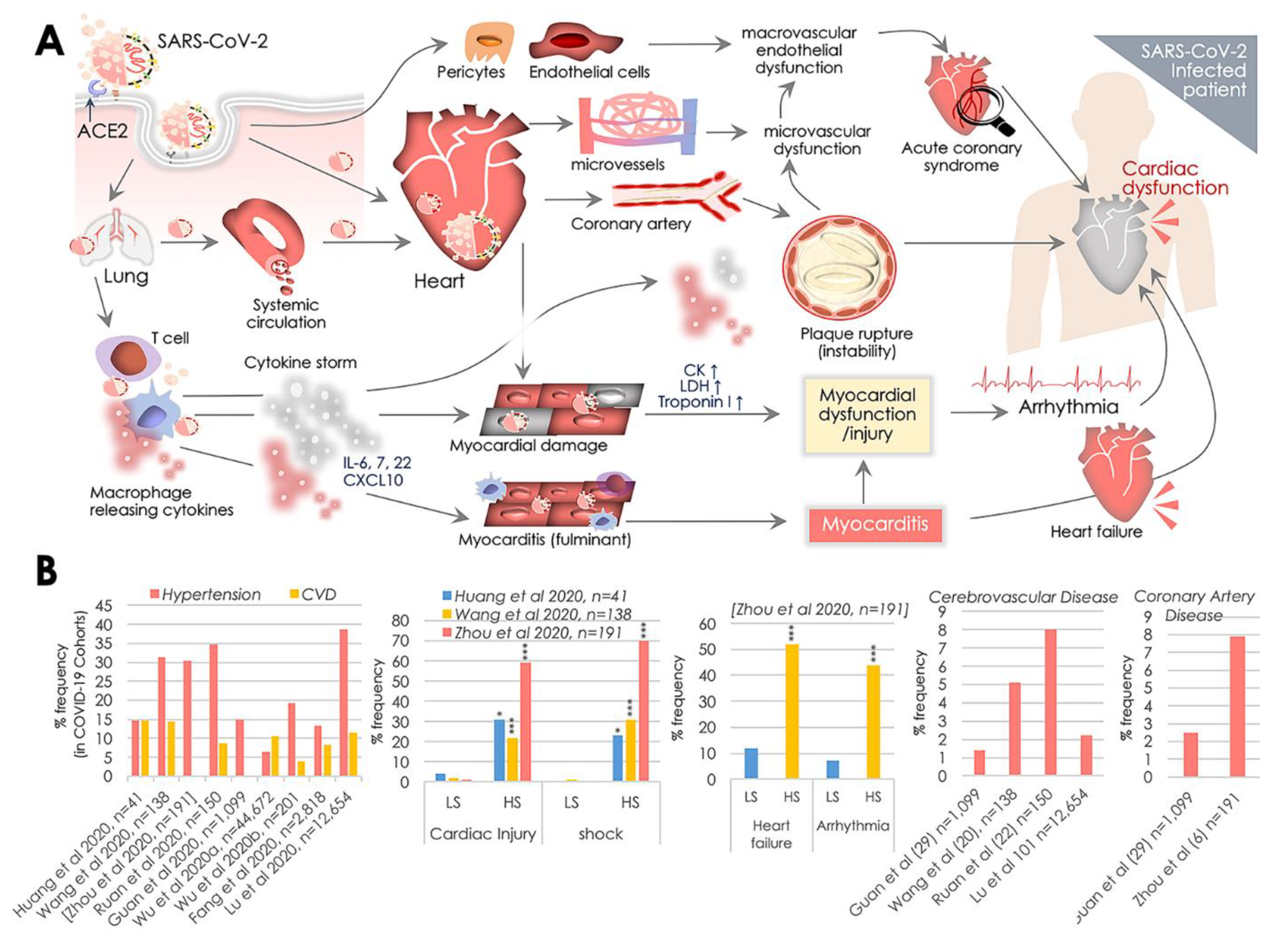
2. SARS-CoV-2, ACE2, and Cardiovascular Challenges
2.1. Myocardial Injury, Shock, and Congestive Cardiac Failure
2.2. Cardiac Arrhythmia
2.3. Myocarditis
2.4. Acute Coronary Disease (ACD) and Ischemia
2.5. Disseminated Intravascular Coagulation (DIC)
2.6. Immune Function in Cardiovascular Complications
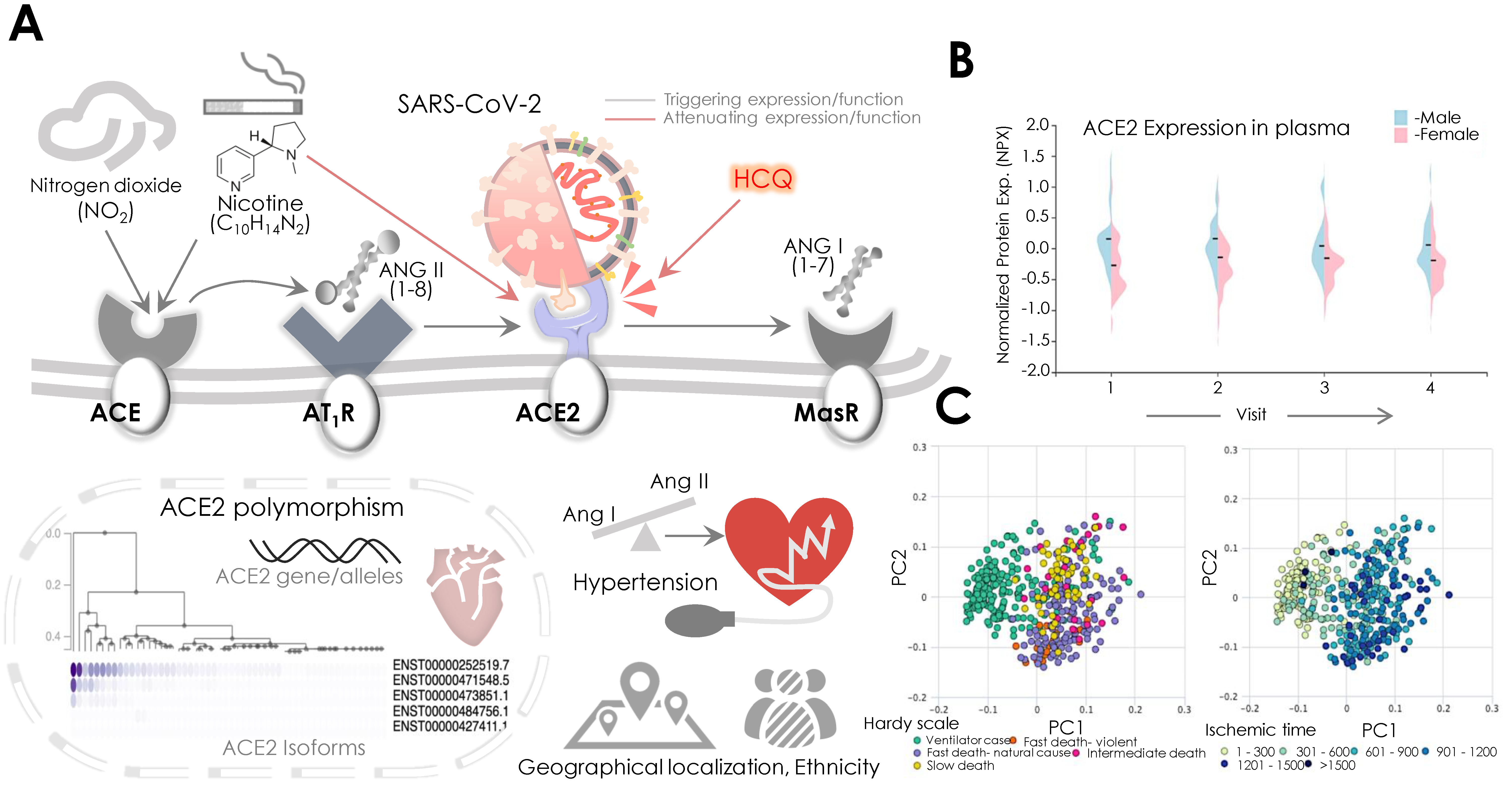
3. ACE2 Receptor and Its Significance in Systemic Cardiovascular Function
4. SARS-CoV-2, ACE2, Hydroxychloroquine and beyond: Preventive and Therapeutic Aspects
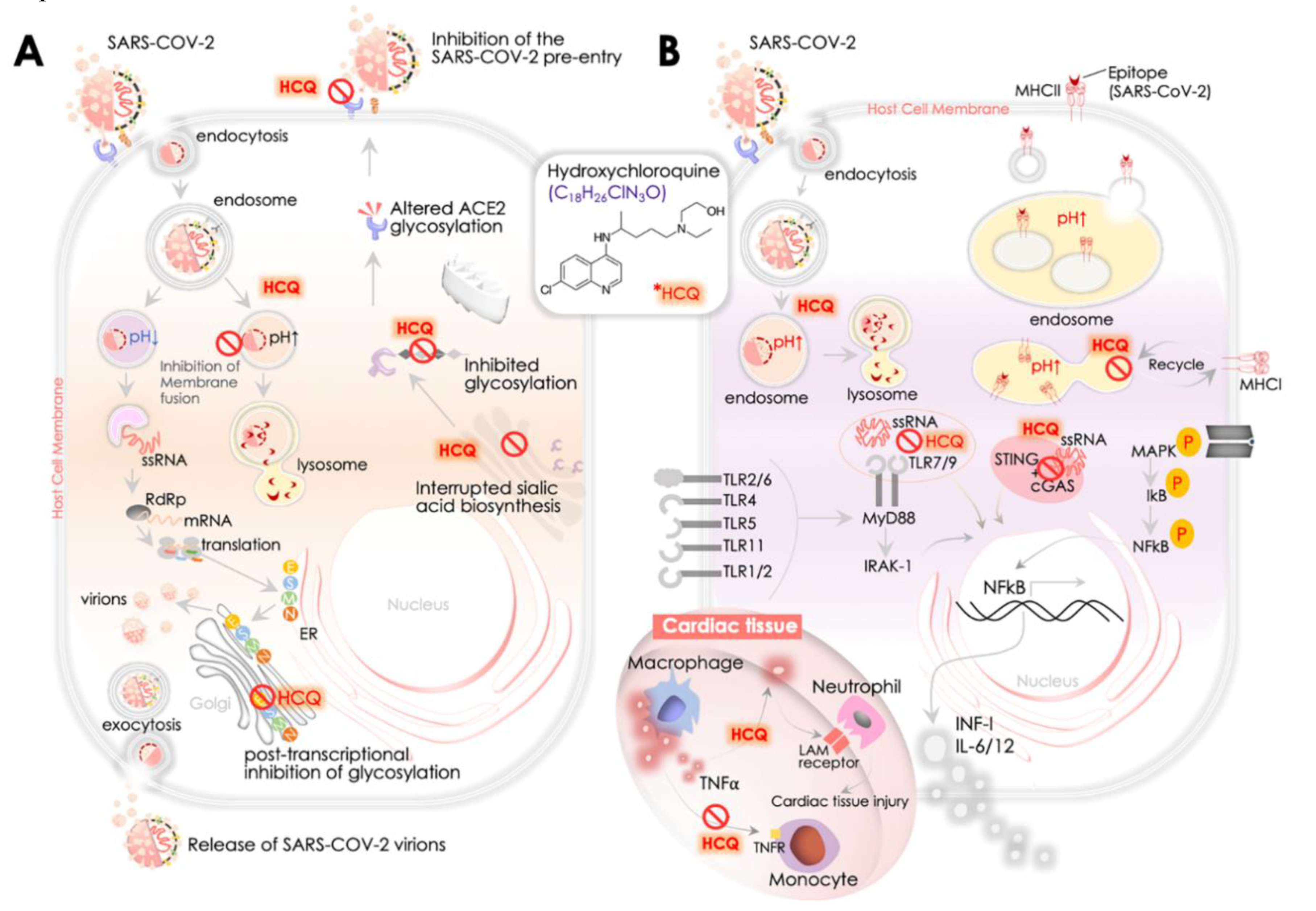
4.1. HCQ Pharmacology, In Vitro and Clinical Outcomes
4.2. ACE2, Hydroxychloroquine, and SARS-CoV-2 Replication
4.3. SARS-CoV-2, ACE2, and HCQ-Mediated Immunomodulatory Response
5. ACE2, HCQ, and Clinical Outcomes: Assessing Cardiovascular Risk and Benefits
5.1. ACE2 and Potential Cardiovascular Risk Factors
5.2. HCQ Therapeutics and its Impact on Cardiovascular Function
5.3. HCQ Repurposing and Heart: Therapeutic Regimes in Current Clinical Trials
6. COVID-19, ACE2, and HCQ: Consideration and Recommendations
7. SARS-CoV-2, ACE2, and HCQ: The Way Forward
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Bondi-Zoccai, G.; Brown, T.S.; Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the Coronavirus Disease 2019 (COVID-19) Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Gomersall, C.D.; Fowler, R.A. Care for Critically Ill Patients with COVID-19. JAMA 2020, 323, 1499–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271.e8–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Keyaerts, E.; Vijgen, L.; Maes, P.; Neyts, J.; Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 2004, 323, 264–268. [Google Scholar] [CrossRef]
- Biot, C.; Daher, W.; Chavain, N.; Fandeur, T.; Khalife, J.; Dive, D.; De Clercq, E. Design and synthesis of hydroxyferroquine derivatives with antimalarial and antiviral activities. J. Med. Chem. 2006, 49, 2845–2849. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020, 57, 279–283. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Liu, L.; Liu, P.; Xu, Q.; Xia, L.; Ling, Y.; Huang, D.; Song, S.; Zhang, D.; et al. [A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19]. Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Med. Sci. 2020, 49, 215–219. [Google Scholar]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.M.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020, 50, 30085–30088. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef] [Green Version]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Sevestre, J.; Mailhe, M.; Doudier, B.; Aubry, C.; Amrane, S.; et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: A pilot observational study. Travel Med. Infect. Dis. 2020, 34, 101663. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.H.; Dhaunsi, G.S.; Makki, B.M.; Qabazard, B.A.; Akhtar, S.; Benter, I.F. Characterization of Angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol. Res. 2012, 66, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1-7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Yi, F.; Wu, K.; Lai, K.; Sun, X.; Zhong, N.; Liu, Z. Clinical Characteristics of 2019 Coronavirus Pneumonia (COVID-19): An Updated Systematic Review. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.03.07.20032573v2 (accessed on 30 June 2020).
- Lu, Y.; Wang, P.; Zhou, T.; Lu, J.; Spatz, E.S.; Nasir, K.; Jiang, L.; Krumholz, H.M. Comparison of Prevalence, Awareness, Treatment, and Control of Cardiovascular Risk Factors in China and the United States. J. Am. Heart Assoc. 2018, 7, e007462. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Fang, Y.Y.; Deng, Y.; Liu, W.; Wang, M.F.; Ma, J.P.; Xiao, W.; Wang, Y.N.; Zhong, M.H.; Li, C.H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med J. 2020, 133, 1025–1031. [Google Scholar] [CrossRef]
- Porcheddu, R.; Serra, C.; Kelvin, D.; Kelvin, N.; Rubino, S. Similarity in Case Fatality Rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J. Infect. Dev. Ctries. 2020, 14, 125–128. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Lavie, C.J.; Sanchis-Gomar, F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): Evidence from a meta-analysis. Prog. Cardiovasc. Dis. 2020. [Google Scholar] [CrossRef]
- Chen, C.F.; Chien, C.H.; Yang, Y.P.; Chou, S.J.; Wang, M.L.; Huo, T.I.; Lin, C.C. Role of Dipeptidyl Peptidase 4 Inhibitors in Diabetic Patients with Coronavirus-19 Infection. J. Chin. Med Assoc. 2020. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musher, D.M.; Abers, M.S.; Corrales-Medina, V.F. Acute Infection and Myocardial Infarction. N. Engl. J. Med. 2019, 380, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Huang, J.; Pan, L. How to balance acute myocardial infarction and COVID-19: The protocols from Sichuan Provincial People’s Hospital. Intensive Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.E.; Park, I.; Ahern, D.J.; Kassiteridi, C.; Danso Abeam, D.; Goddard, M.E.; Green, P.; Maffia, P.; Monaco, C. Immune cell census in murine atherosclerosis: Cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc. Res. 2018, 114, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Heart J. 2020, 41, 1858. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Ziegler, L.; Gajulapuri, A.; Frumento, P.; Bonomi, A.; Wallen, H.; de Faire, U.; Rose-John, S.; Gigante, B. Interleukin 6 trans-signalling and risk of future cardiovascular events. Cardiovasc. Res. 2019, 115, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, G.; Condorelli, G. Interleukin-6 trans-signalling and risk of future cardiovascular events: A new avenue for atheroprotection? Cardiovasc. Res. 2019, 115, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: The good, bad, and absolute? American journal of physiology. Heart Circ. Physiol. 2016, 310, H137–H152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostal, D.E.; Baker, K.M. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ. Res. 1999, 85, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Chappell, M.C. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: More than regulation of blood pressure? Hypertension 2007, 50, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Lazartigues, E. Angiotensin-converting enzyme 2: Central regulator for cardiovascular function. Curr. Hypertens. Rep. 2010, 12, 170–175. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Gu, H.; Zhao, Z.; Wang, W.; Cao, B.; Lai, C.; Yang, X.; Zhang, L.; Duan, Y.; Zhang, S.; et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci. Rep. 2014, 4, 7027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yang, N.; Tang, J.; Liu, S.; Luo, D.; Duan, Q.; Wang, X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014, 185, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.R.; Scholey, J.W. Angiotensin-converting enzyme 2 and renal disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 35–41. [Google Scholar] [CrossRef]
- Assiri, A.; McGeer, A.; Perl, T.M.; Price, C.S.; Al Rabeeah, A.A.; Cummings, D.A.; Alabdullatif, Z.N.; Assad, M.; Almulhim, A.; Makhdoom, H.; et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 2013, 369, 407–416. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in coronavirus disease 2019 patients: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Simoes, E.S.A.C.; Teixeira, M.M. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol. Res. 2016, 107, 154–162. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Y.X.; Zhang, Y.H.; Zhu, L.; Deng, B.P.; Zhou, Z.L.; Li, S.Y.; Lu, X.T.; Song, L.L.; Lei, X.M.; et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15886–15891. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Parajuli, N.; Ramprasath, T.; Patel, V.B.; Wang, W.; Putko, B.; Mori, J.; Oudit, G.Y. Targeting angiotensin-converting enzyme 2 as a new therapeutic target for cardiovascular diseases. Can. J. Physiol. Pharmacol. 2014, 92, 558–565. [Google Scholar] [CrossRef]
- Khanna, A.; English, S.W.; Wang, X.S.; Ham, K.; Tumlin, J.; Szerlip, H.; Busse, L.W.; Altaweel, L.; Albertson, T.E.; Mackey, C.; et al. Angiotensin II for the Treatment of Vasodilatory Shock. N. Engl. J. Med. 2017, 377, 419–430. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pohlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pohlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Coatney, G.R. Pitfalls in a discovery: The chronicle of chloroquine. Am. J. Trop. Med. Hyg. 1963, 12, 121–128. [Google Scholar] [CrossRef]
- Raoult, D.; Drancourt, M.; Vestris, G. Bactericidal effect of doxycycline associated with lysosomotropic agents on Coxiella burnetii in P388D1 cells. Antimicrob. Agents Chemother. 1990, 34, 1512–1514. [Google Scholar] [CrossRef] [Green Version]
- Boulos, A.; Rolain, J.M.; Raoult, D. Antibiotic susceptibility of Tropheryma whipplei in MRC5 cells. Antimicrob. Agents Chemother. 2004, 48, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Rolain, J.M.; Colson, P.; Raoult, D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 2007, 30, 297–308. [Google Scholar] [CrossRef]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? The Lancet. Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Boelaert, J.R.; Piette, J.; Sperber, K. The potential place of chloroquine in the treatment of HIV-1-infected patients. J. Clin. Virol. 2001, 20, 137–140. [Google Scholar] [CrossRef]
- Lee, S.J.; Silverman, E.; Bargman, J.M. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat. Rev. Nephrol. 2011, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Blau, D.M.; Holmes, K.V. Human coronavirus HCoV-229E enters susceptible cells via the endocytic pathway. Adv. Exp. Med. Biol. 2001, 494, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Tatsumi, K.; Imai, A.M.; Saito, K.; Kuriyama, T.; Shirasawa, H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antivir. Res. 2008, 77, 150–152. [Google Scholar] [CrossRef]
- Keyaerts, E.; Li, S.; Vijgen, L.; Rysman, E.; Verbeeck, J.; Van Ranst, M.; Maes, P. Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob. Agents Chemother. 2009, 53, 3416–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winzeler, E.A. Malaria research in the post-genomic era. Nature 2008, 455, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, S.; Poole, B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J. Cell Biol. 1981, 90, 656–664. [Google Scholar] [CrossRef]
- Popert, A.J. Chloroquine: A review. Rheumatol. Rehabil. 1976, 15, 235–238. [Google Scholar] [CrossRef]
- Lim, H.S.; Im, J.S.; Cho, J.Y.; Bae, K.S.; Klein, T.A.; Yeom, J.S.; Kim, T.S.; Choi, J.S.; Jang, I.J.; Park, J.W. Pharmacokinetics of hydroxychloroquine and its clinical implications in chemoprophylaxis against malaria caused by Plasmodium vivax. Antimicrob. Agents Chemother. 2009, 53, 1468–1475. [Google Scholar] [CrossRef] [Green Version]
- Andreani, J.; Le Bideau, M.; Duflot, I.; Jardot, P.; Rolland, C.; Boxberger, M.; Wurtz, N.; Rolain, J.M.; Colson, P.; La Scola, B.; et al. In vitro testing of combined hydroxychloroquine and azithromycin on SARS-CoV-2 shows synergistic effect. Microb. Pathog. 2020, 145, 104228. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, J.; Zhang, Z.; Jiang, S.; Han, S.; Yan, D.; Zhuang, R.; Hu, B.; Zhang, Z. Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. MedRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.03.22.20040758v3 (accessed on 30 June 2020).
- Mahevas, M.; Tran, V.-T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Gallien, S.; Lepeule, R.; Szwebel, T.-A.; Lescure, X. No evidence of clinical efficacy of hydroxychloroquine in patients hospitalized for COVID-19 infection with oxygen requirement: Results of a study using routinely collected data to emulate a target trial. MedRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.10.20060699v1 (accessed on 30 June 2020).
- Tang, W.; Cao, Z.; Han, M.; Wang, Z.; Chen, J.; Sun, W.; Wu, Y.; Xiao, W.; Liu, S.; Chen, E.; et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: Open label, randomised controlled trial. BMJ 2020, 369, m1849. [Google Scholar] [CrossRef]
- Million, M.; Lagier, J.C.; Gautret, P.; Colson, P.; Fournier, P.E.; Amrane, S.; Hocquart, M.; Mailhe, M.; Esteves-Vieira, V.; Doudier, B.; et al. Early treatment of COVID-19 patients with hydroxychloroquine and azithromycin: A retrospective analysis of 1061 cases in Marseille, France. Travel Med. Infect. Dis. 2020, 35, 101738. [Google Scholar] [CrossRef] [PubMed]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. MedRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.16.20065920v2 (accessed on 30 June 2020).
- Mathies, D.; Rauschning, D.; Wagner, U.; Mueller, F.; Maibaum, M.; Binnemann, C.; Waldeck, S.; Thinnes, K.; Braun, M.; Schmidbauer, W.; et al. A Case of SARS-CoV-2-pneumonia with successful antiviral therapy in a 77-year-old male with heart transplant. Am. J. Transplant. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.C.E.; Weaver, J.; Kostka, K.; Duarte-Salles, T.; Abrahao, M.T.F.; Alghoul, H.; Alser, O.; Alshammari, T.M.; Biedermann, P.; Burn, E. Safety of hydroxychloroquine, alone and in combination with azithromycin, in light of rapid wide-spread use for COVID-19: A multinational, network cohort and self-controlled case series study. MedRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.08.20054551v2 (accessed on 30 June 2020).
- Kwiek, J.J.; Haystead, T.A.; Rudolph, J. Kinetic mechanism of quinone oxidoreductase 2 and its inhibition by the antimalarial quinolines. Biochemistry 2004, 43, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Sialic acids as ligands in recognition phenomena. FASEB J. 1997, 11, 248–255. [Google Scholar] [CrossRef]
- Olofsson, S.; Kumlin, U.; Dimock, K.; Arnberg, N. Avian influenza and sialic acid receptors: More than meets the eye? Lancet Infect. Dis. 2005, 5, 184–188. [Google Scholar] [CrossRef]
- Byrd, T.F.; Horwitz, M.A. Chloroquine inhibits the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. A potential new mechanism for the therapeutic effect of chloroquine against intracellular pathogens. J. Clin. Investig. 1991, 88, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Legssyer, R.; Josse, C.; Piette, J.; Ward, R.J.; Crichton, R.R. Changes in function of iron-loaded alveolar macrophages after in vivo administration of desferrioxamine and/or chloroquine. J. Inorg. Biochem. 2003, 94, 36–42. [Google Scholar] [CrossRef]
- Gay, B.; Bernard, E.; Solignat, M.; Chazal, N.; Devaux, C.; Briant, L. pH-dependent entry of chikungunya virus into Aedes albopictus cells. Infect. Genet. Evol. 2012, 12, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Tricou, V.; Minh, N.N.; Van, T.P.; Lee, S.J.; Farrar, J.; Wills, B.; Tran, H.T.; Simmons, C.P. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis. 2010, 4, e785. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Santhosh, S.R.; Tiwari, M.; Lakshmana Rao, P.V.; Parida, M. Assessment of in vitro prophylactic and therapeutic efficacy of chloroquine against Chikungunya virus in vero cells. J. Med. Virol. 2010, 82, 817–824. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, L.; Rutala, W.A.; Weber, D.J.; Sobsey, M.D. Coronavirus survival on healthcare personal protective equipment. Infect. Control Hosp. Epidemiol. 2010, 31, 560–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, N.E. Examination of potential inhibitors of hepatitis A virus uncoating. Intervirology 1998, 41, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Gennero, L.; Sperber, K.; Boelaert, J.R. The anti-HIV-1 activity of chloroquine. J. Clin. Virol. 2001, 20, 131–135. [Google Scholar] [CrossRef]
- Fonseca, B.A.; Pincus, S.; Shope, R.E.; Paoletti, E.; Mason, P.W. Recombinant vaccinia viruses co-expressing dengue-1 glycoproteins prM and E induce neutralizing antibodies in mice. Vaccine 1994, 12, 279–285. [Google Scholar] [CrossRef]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef] [Green Version]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994, 68, 6523–6534. [Google Scholar] [CrossRef] [Green Version]
- Perrier, A.; Bonnin, A.; Desmarets, L.; Danneels, A.; Goffard, A.; Rouille, Y.; Dubuisson, J.; Belouzard, S. The C-terminal domain of the MERS coronavirus M protein contains a trans-Golgi network localization signal. J. Biol. Chem. 2019, 294, 14406–14421. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.H.; Choi, J.H.; Byun, M.S.; Jue, D.M. Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology 2006, 45, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Seitz, M.; Valbracht, J.; Quach, J.; Lotz, M. Gold sodium thiomalate and chloroquine inhibit cytokine production in monocytic THP-1 cells through distinct transcriptional and posttranslational mechanisms. J. Clin. Immunol. 2003, 23, 477–484. [Google Scholar] [CrossRef]
- Zhu, X.; Ertel, W.; Ayala, A.; Morrison, M.H.; Perrin, M.M.; Chaudry, I.H. Chloroquine inhibits macrophage tumour necrosis factor-alpha mRNA transcription. Immunology 1993, 80, 122–126. [Google Scholar] [PubMed]
- Farias, K.J.; Machado, P.R.; de Almeida Junior, R.F.; de Aquino, A.A.; da Fonseca, B.A. Chloroquine interferes with dengue-2 virus replication in U937 cells. Microbiol. Immunol. 2014, 58, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Jue, D.M. Chloroquine inhibits processing of tumor necrosis factor in lipopolysaccharide-stimulated RAW 264.7 macrophages. J. Immunol. 1997, 158, 4901–4907. [Google Scholar]
- Bondeson, J.; Sundler, R. Antimalarial drugs inhibit phospholipase A2 activation and induction of interleukin 1beta and tumor necrosis factor alpha in macrophages: Implications for their mode of action in rheumatoid arthritis. Gen. Pharmacol. 1998, 30, 357–366. [Google Scholar] [CrossRef]
- van den Borne, B.E.; Dijkmans, B.A.; de Rooij, H.H.; le Cessie, S.; Verweij, C.L. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J. Rheumatol. 1997, 24, 55–60. [Google Scholar]
- Karres, I.; Kremer, J.P.; Dietl, I.; Steckholzer, U.; Jochum, M.; Ertel, W. Chloroquine inhibits proinflammatory cytokine release into human whole blood. Am. J. Physiol. 1998, 274, R1058–R1064. [Google Scholar] [CrossRef]
- Nooteboom, A.; Hendriks, T.; Otteholler, I.; van der Linden, C.J. Permeability characteristics of human endothelial monolayers seeded on different extracellular matrix proteins. Mediat. Inflamm. 2000, 9, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.Y.; Choi, J.W.; Jeon, K.I.; Jue, D.M. Chloroquine decreases cell-surface expression of tumour necrosis factor receptors in human histiocytic U-937 cells. Immunology 2002, 105, 83–91. [Google Scholar] [CrossRef]
- Wolfe, F.; Caplan, L.; Michaud, K. Treatment for rheumatoid arthritis and the risk of hospitalization for pneumonia: Associations with prednisone, disease-modifying antirheumatic drugs, and anti-tumor necrosis factor therapy. Arthritis Rheum. 2006, 54, 628–634. [Google Scholar] [CrossRef]
- Bernatsky, S.; Hudson, M.; Suissa, S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology 2007, 46, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Smitten, A.L.; Choi, H.K.; Hochberg, M.C.; Suissa, S.; Simon, T.A.; Testa, M.A.; Chan, K.A. The risk of hospitalized infection in patients with rheumatoid arthritis. J. Rheumatol. 2008, 35, 387–393. [Google Scholar]
- Rokni, M.; Ghasemi, V.; Tavakoli, Z. Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS. Rev. Med Virol. 2020, 30, e2107. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.; Morrison, T.E.; Funkhouser, W.; Uematsu, S.; Akira, S.; Baric, R.S.; Heise, M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008, 4, e1000240. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [Green Version]
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. 2014, 25, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Lokugamage, K.G.; Hage, A.; Schindewolf, C.; Rajsbaum, R.; Menachery, V.D. SARS-CoV-2 sensitive to type I interferon pretreatment. BioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.03.07.982264v3 (accessed on 30 June 2020).
- Austin Taylor, M.; Bennett, M.; Kumar, V.; Schatzle, J.D. Functional defects of NK cells treated with chloroquine mimic the lytic defects observed in perforin-deficient mice. J. Immunol. 2000, 165, 5048–5053. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yang, X.; Yang, J.; Li, M. Hydroxychloroquine Inhibits the Differentiation of Th17 Cells in Systemic Lupus Erythematosus. J. Rheumatol. 2018, 45, 818–826. [Google Scholar] [CrossRef]
- Oh, S.; Shin, J.H.; Jang, E.J.; Won, H.Y.; Kim, H.K.; Jeong, M.G.; Kim, K.S.; Hwang, E.S. Anti-inflammatory activity of chloroquine and amodiaquine through p21-mediated suppression of T cell proliferation and Th1 cell differentiation. Biochem. Biophys. Res. Commun. 2016, 474, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Schrezenmeier, E.; Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef]
- Mocholi, E.; Dowling, S.D.; Botbol, Y.; Gruber, R.C.; Ray, A.K.; Vastert, S.; Shafit-Zagardo, B.; Coffer, P.J.; Macian, F. Autophagy Is a Tolerance-Avoidance Mechanism that Modulates TCR-Mediated Signaling and Cell Metabolism to Prevent Induction of T Cell Anergy. Cell Rep. 2018, 24, 1136–1150. [Google Scholar] [CrossRef] [Green Version]
- Briant, L.; Robert-Hebmann, V.; Acquaviva, C.; Pelchen-Matthews, A.; Marsh, M.; Devaux, C. The protein tyrosine kinase p56lck is required for triggering NF-kappaB activation upon interaction of human immunodeficiency virus type 1 envelope glycoprotein gp120 with cell surface CD4. J. Virol. 1998, 72, 6207–6214. [Google Scholar] [CrossRef] [Green Version]
- Kreutz, R.; Algharably, E.A.E.; Azizi, M.; Dobrowolski, P.; Guzik, T.; Januszewicz, A.; Persu, A.; Prejbisz, A.; Riemer, T.G.; Wang, J.G.; et al. Hypertension, the renin-angiotensin system, and the risk of lower respiratory tract infections and lung injury: Implications for COVID-19. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, F.; Masia, M.; Mirete, C.; Soldan, B.; Rodriguez, J.C.; Padilla, S.; Hernandez, I.; Royo, G.; Martin-Hidalgo, A. The influence of age and gender on the population-based incidence of community-acquired pneumonia caused by different microbial pathogens. J. Infect. 2006, 53, 166–174. [Google Scholar] [CrossRef]
- Soto, M.; Bang, S.I.; McCombs, J.; Rodgers, K.E. Renin Angiotensin system-modifying therapies are associated with improved pulmonary health. Clin. Diabetes Endocrinol. 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.; Zaizafoun, M.; Stock, E.; Ghamande, S.; Arroliga, A.C.; White, H.D. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Proceedings 2018, 31, 419–423. [Google Scholar] [CrossRef]
- Sanchez-Aguilar, M.; Ibarra-Lara, L.; Del Valle-Mondragon, L.; Rubio-Ruiz, M.E.; Aguilar-Navarro, A.G.; Zamorano-Carrillo, A.; Ramirez-Ortega, M.D.C.; Pastelin-Hernandez, G.; Sanchez-Mendoza, A. Rosiglitazone, a Ligand to PPARgamma, Improves Blood Pressure and Vascular Function through Renin-Angiotensin System Regulation. PPAR Res. 2019, 2019, 1371758. [Google Scholar] [CrossRef] [Green Version]
- Beaney, T.; Burrell, L.M.; Castillo, R.R.; Charchar, F.J.; Cro, S.; Damasceno, A.; Kruger, R.; Nilsson, P.M.; Prabhakaran, D.; Ramirez, A.J.; et al. May Measurement Month 2018: A pragmatic global screening campaign to raise awareness of blood pressure by the International Society of Hypertension. Eur. Heart J. 2019, 40, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Meulenbelt, J.; van Bree, L.; Dormans, J.A.; Boink, A.B.; Sangster, B. Biochemical and histological alterations in rats after acute nitrogen dioxide intoxication. Hum. Exp. Toxicol. 1992, 11, 189–200. [Google Scholar] [CrossRef]
- Patel, J.M.; Sekharam, K.M.; Block, E.R. Oxidant injury increases cell surface receptor binding of angiotensin II to pulmonary artery endothelial cells. J. Biochem. Toxicol. 1990, 5, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.M.; Fuchs, R.M.; Gardner, J.D.; Lazartigues, E.; Yue, X. Nicotine and the renin-angiotensin system. American journal of physiology. Regul. Integr. Comp. Physiol. 2018, 315, R895–R906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippi, G.; Henry, B.M. Active smoking is not associated with severity of coronavirus disease 2019 (COVID-19). Eur. J. Intern. Med. 2020, 75, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Guo, Y.; Markevych, I.; Qian, Z.M.; Bloom, M.S.; Heinrich, J.; Dharmage, S.C.; Rolling, C.A.; Jordan, S.S.; Komppula, M.; et al. Association of Long-term Exposure to Ambient Air Pollutants With Risk Factors for Cardiovascular Disease in China. JAMA Netw. Open 2019, 2, e190318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zhang, Z.F.; Froines, J.; Zhao, J.; Wang, H.; Yu, S.Z.; Detels, R. Air pollution and case fatality of SARS in the People’s Republic of China: An ecologic study. Environ. Health Glob. Access Sci. Source 2003, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cong, M.; Wang, N.; Li, X.; Zhang, H.; Zhang, K.; Jin, M.; Wu, N.; Qiu, C.; Li, J. Association of angiotensin-converting enzyme 2 gene polymorphism and enzymatic activity with essential hypertension in different gender: A case-control study. Medicine 2018, 97, e12917. [Google Scholar] [CrossRef]
- Lu, N.; Yang, Y.; Wang, Y.; Liu, Y.; Fu, G.; Chen, D.; Dai, H.; Fan, X.; Hui, R.; Zheng, Y. ACE2 gene polymorphism and essential hypertension: An updated meta-analysis involving 11,051 subjects. Mol. Biol. Rep. 2012, 39, 6581–6589. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Lu, X.; Zhao, Q.; Rao, D.C.; Jaquish, C.E.; Hixson, J.E.; Chen, J.; Wang, L.; Cao, J.; et al. Polymorphisms of ACE2 are associated with blood pressure response to cold pressor test: The GenSalt study. Am. J. Hypertens. 2012, 25, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, D.S.; Santos, R.S.; Jardim, P.; Silva, E.G.; Reis, A.A.S.; Pedrino, G.R.; Ulhoa, C.J. The combination of ACE I/D and ACE2 G8790A polymorphisms revels susceptibility to hypertension: A genetic association study in Brazilian patients. PLoS ONE 2019, 14, e0221248. [Google Scholar] [CrossRef] [Green Version]
- Schouten, L.R.; van Kaam, A.H.; Kohse, F.; Veltkamp, F.; Bos, L.D.; de Beer, F.M.; van Hooijdonk, R.T.; Horn, J.; Straat, M.; Witteveen, E.; et al. Age-dependent differences in pulmonary host responses in ARDS: A prospective observational cohort study. Ann. Intensive Care 2019, 9, 55. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Touret, F.; de Lamballerie, X. Of chloroquine and COVID-19. Antivir. Res. 2020, 177, 104762. [Google Scholar] [CrossRef] [PubMed]
- Calain, P. The Ebola clinical trials: A precedent for research ethics in disasters. J. Med. Ethics 2018, 44, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisk-Holmberg, M.; Bergqvist, Y.; Englund, U. Chloroquine intoxication. Br. J. Clin. Pharmacol. 1983, 15, 502–503. [Google Scholar] [CrossRef]
- Chatre, C.; Roubille, F.; Vernhet, H.; Jorgensen, C.; Pers, Y.M. Cardiac Complications Attributed to Chloroquine and Hydroxychloroquine: A Systematic Review of the Literature. Drug Saf. 2018, 41, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Laing, R.; Waning, B.; Gray, A.; Ford, N.; t Hoen, E. 25 years of the WHO essential medicines lists: Progress and challenges. Lancet 2003, 361, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Crumb, W.J., Jr.; Vicente, J.; Johannesen, L.; Strauss, D.G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 2016, 81, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnesmann, E.; Kandolf, R.; Lewalter, T. Chloroquine cardiomyopathy—A review of the literature. Immunopharmacol. Immunotoxicol. 2013, 35, 434–442. [Google Scholar] [CrossRef]
- Mzayek, F.; Deng, H.; Mather, F.J.; Wasilevich, E.C.; Liu, H.; Hadi, C.M.; Chansolme, D.H.; Murphy, H.A.; Melek, B.H.; Tenaglia, A.N.; et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2007, 2, e6. [Google Scholar] [CrossRef] [PubMed]
- Chorin, E.; Wadhwani, L.; Magnani, S.; Dai, M.; Shulman, E.; Nadeau-Routhier, C.; Knotts, R.; Bar-Cohen, R.; Kogan, E.; Barbhaiya, C. QT Interval Prolongation and Torsade De Pointes in Patients with COVID-19 treated with Hydroxychloroquine/Azithromycin. Heart Rhythm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Geier, M.; Marzi, A.; Krumbiegel, M.; Peipp, M.; Fey, G.H.; Gramberg, T.; Pohlmann, S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 2004, 319, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asan Medical Center. Comparison of Lopinavir/Ritonavir or Hydroxychloroquine in Patients with Mild Coronavirus Disease (COVID-19). Available online: https://ClinicalTrials.gov/show/NCT04307693 (accessed on 30 June 2020).
- Shanghai Public Health Clinical Center. Efficacy and Safety of Hydroxychloroquine for Treatment of COVID-19. Available online: https://ClinicalTrials.gov/show/NCT04261517 (accessed on 30 June 2020).
- University of Minnesota; McGill University Health Centre/Research Institute of the McGill University Health Centre; University of Manitoba; University of Alberta. Post-exposure Prophylaxis Preemptive Therapy for SARS-Coronavirus-2. Available online: https://ClinicalTrials.gov/show/NCT04308668 (accessed on 30 June 2020).
- Corrales-Medina, V.F.; Alvarez, K.N.; Weissfeld, L.A.; Angus, D.C.; Chirinos, J.A.; Chang, C.C.; Newman, A.; Loehr, L.; Folsom, A.R.; Elkind, M.S.; et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 2015, 313, 264–274. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, L.; Sun, X.; Yan, Z.; Hu, C.; Wu, J.; Xu, L.; Li, X.; Liu, H.; Yin, P.; et al. Altered Lipid Metabolism in Recovered SARS Patients Twelve Years after Infection. Sci. Rep. 2017, 7, 9110. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Investigation/References | Cell Systems | Drug, Concentration, and Assay Time (h) | Study Control | Key Findings/Comments |
|---|---|---|---|---|
| Yao et al. 2020 | Vero E6 cell (Origin-African green Monkey) | CQ and HCQ 0.032, 0.16, 0.80, 4, 20, & 100 µM 2 h | - | -HCQ showed better SARS-CoV-2 inhibitory activity than CQ. -An extended incubation period may produce greater anti-viral effect |
| Liu et al. 2020 | Vero E6 Cells | CQ and HCQ 0.068, 0.21, 0.62, 1.85, 5.56, 16.67, and 50 µM 1 h | PBS (Phosphate buffer saline) | -HCQ inhibited the steps including infection/entry and post-infection -At the higher viral replication rate, anti-viral efficacy of HCQ found to be lesser than of CQ |
| Wang et al. 2020 | Vero E6 Cells | CQ and others * 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 µM 1 h | DMSO | -HCQ inhibited the viral activity at low µM conc. (effective conc. EC50 = 1.13 μM) -CQ effectively inhibited SARS-CoV-2 infection in vitro |
| Andreani et al. 2020 | Vero E6 cells | CQ- 1, 2 or 5 μM associated with 5 or 10 μM for azithromycin. | - | Combination of hydroxychloroquine and azithromycin has a synergistic effect in vitro on SARS-CoV-2 at concentrations |
| Keyaerts et al. 2004 (*Earliest report from the SARS-CoV) | Vero E6 cell | CQ 0, 0.8, 4, 20, & 100 µM 8 h to 3 days | - | -CQ potently inhibits SARS-CoV activity at a lesser (8.8 ± 1.2 μM) concentration than its cytostatic activity (261.3 ± 14.5 μM) -Addition of CQ even after 5 h of SARS-CoV infection could yet be inhibitory active |
| Investigation/Reference | Investigation Type/Design | Patients (Total No) | Regimes | Severity of COVID-19 Disease | Results/Key Findings | Comment | Location | Limitation | |
|---|---|---|---|---|---|---|---|---|---|
| Con | HCQ | ||||||||
| Chen J et al. (2020) | Randomized and controlled trial | 15 | 15 | HCQ- 400 mg for 5 days | 6–7 days symptomatic patients, unclear severity | Indifferent outcomes in groups. By day 7, no significant change in conversion rate (86.7% vs 93.3%) observed. | Patients were tested negative for COVID-19 at 2 weeks | Shanghai, China | Smaller sample size. Not peer-reviewed, availability in Chinese language |
| Gautret P et al. (2020a) | Open-label trail, Non-randomized, Non-blinded | 16 | 26 | HCQ- 600 mg for 10 days | Asymptomatic patients-17%, Patients with respiratory symptoms- 61%, Chest CT pneumonia +ve patients- 22% | Unadjusted results showed significantly reduced viral titer at day 6 (HCQ-70% vs. con 12.5%, PCR based, p < 0.01) | Exclusion of 6 patients from data (1- died, 1- withdrew, 3 needed ICU admission, 1- lost follow-up) | Marseille, France | Study design, Smaller sample-size, Exclusion of 6 patients, inconclusive long-term outcomes |
| Molina JM et al. (2020) | Prospective open-label investigation | 0 | 10 | HCQ- 600 mg for 5 days + AZM 500 mg × 1, then 250 mg | 10 patients out of 11 were on supplemental oxygen | 8 patients out of 10 were positive at day 5–6 (nasopharyngeal swab) (80%, 95% CI: 49–94) | Patient died-1, Patient transferred to ICU-2, Patient had no further HCQ post prolongation of QTc-1 | Paris, France | Smaller sample size. Not peer-reviewed. |
| Chen Z et al. (2020) | Parallel-group trail Randomized | 31 | 31 | HCQ- 400 mg for 5 days | Mild illness was observed in CT confirmed pneumonia cases | - Clinical recovery and cough remission time reduced in HCQ group, while resolution of pneumonia was higher (80.60% vs. 54.8%) in the HCQ group. | Undefined status, 4 patients developed severe illness in the control group | Wuhan, China | Smaller sample size. Not peer-reviewed. |
| Gautret P et al. (2020b) | Open-label trail, Non-randomized, Non-blinded | 0 | 80 | HCQ- 600 mg for 10 days + 500 mg, followed by 250 mg AZM | Asymptomatic- 5%, Pneumonia cases- 54%, Patients with low national early warning score (NEWS) and mild disease- 92% | Decreased nasopharyngeal viral load at 7th (83% negative) and 8th (93%) days | Patients discharged from hospital - 65 (81.3%), Patients needed ICU admission- 1, Deceased- 1 | Marseille, France | Design of the study, Smaller sample size. Not peer-reviewed. Short follow-up time period |
| Tang W et al. (2020) | Open-label, Multi-centric, Randomized, Controlled trial | 75 | 75 | HCQ- 200 mg for first 3 days, 800 mg for remaining days (total 2–3 weeks) | Patients with mild-moderate disease- 148. Patients with severe illness-2 | HCQ showed no significantly higher negative conversion probability (85.4%) than control (81.3%) patients. Adverse effects were reported in HCQ group | Adverse events in control and HCQ group were reported in 7 and 21 patients respectively | Shanghai, Anhui, Hubei, China | Smaller sample size. Not peer-reviewed. |
| Million M et al. (2020) | Open-label trail, Non-randomized, Non-blinded | 0 | 1061 | HCQ- 200 mg (3 X/day) for 10 days + 500 mg AZM (day-1), followed by 250 mg for next 4 days | Patients had 20.5% and 2.2% moderate and severity scores respectively | In 10 day regime, good clinical results and virological cure were reported in 973 patients (91.7%). HCQ+AZM treatment before COVID-19 illness is safe and has low fatality rate in patients | Majority of patients had relatively mild symptoms at start (95%), therefore, only 10 patients (0.9%) transferred to the ICU, & 8 (0.75%) patients died | Marseille, France | Study design. Incomplete data on some patients. Unsynchronized diagnostic reports |
| Mahevas M et al. (2020) | Multi-centric, Non-Randomized, aim to emulate a target trial | 97 | 84 | HCQ- 600 mg for about ~7–8 days | Most patients had bilateral pneumonia, and 75% moderate or severe illness | No significant relief was observed in HCQ group as compared to control at day 7 in hospitalized patients. All comorbidities were less frequent in the HCQ group. | 17 (20%) patients in the HCQ group, received concomitant AZM, while 64 (76%) received amoxicillin and clavulanic acid. | Créteil, Suresnes, Evry, and Paris, France | Not peer-reviewed. No randomization, Unbalanced prognostic variables across hospitals. |
| Magagnoli J et al. (2020) | Retrospective analysis, Non-randomized | 158 | 97 (HCQ), 113 (HCQ+AZ) | - | All confirmed COVID-19 patients. No severity was specified | No evidence of HCQ either with or without AZM, lessen the risk of mechanical support in patients | Study comprises only men aged over 65 years, most black population | Virginia, and South Carolina, USA | Study design. Not peer-reviewed. Possibility of selection bias. |
| Mathies D et al. (2020) -Case report | Case report | 0 | 1 | HCQ- 400 mg for 1st day, then 200 mg for remaining 11 days | 77-year-old COVID-19 positive patient with a heart transplant, moderate symptoms | Patient with existing dyspnea and dry cough, showed no further deterioration of the clinical state post HCQ medication. After 12 days, all negative | Patients survived and discharged from hospital after 12 days and had symptoms | Koblenz, Germany | - |
| Lane JCE et al. (2020) -Case series | A multinational, network cohort and self-controlled case series study | 310, 350 (SSZ) | HCQ-956374 HCQ+AZM- 323122, HCQ+ AMX- 351956 | - (variable) | 16 patients had severe adverse events | No excess risk of severe events was identified when 30-day HCQ and SSZ (sulfasalazine) were compare. While, AZM + HCQ increased risk CVD and morality | cardiovascular complications in HCQ+AZM group are likely due to synergistic effects on QT length | Germany, Japan, USA Netherlands, Spain, & UK. | Not peer-reviewed. Potential risk of overlapping in patient datasets, variance in data |
| Trail Identifier | Study Title | Study Type/Design | Study Phase | Volunteers (Active) | Interventions/Drug(s) | Active Comparator | Primary Outcome | Location | Study Sponsor |
|---|---|---|---|---|---|---|---|---|---|
| NCT04371926 | Prophylactic Benefit of HCQ in COVID-19 Cases with Mild to Moderate Symptoms and in Healthcare Workers with High Exposure Risk (PREVENT) | Interventional, Randomized | - | 64 | HCQ, 400 mg (day-1), then 200 mg for next 4 days (b.i.d.) | No-HCQ arm | Prophylactic Benefit of HCQ in patients and healthcare workers | - | Texas Cardiac Arrhythmia Research Foundation |
| NCT04341441 | Will Hydroxychloroquine Impede or Prevent COVID-19 (WHIP COVID-19) | Interventional, Randomized | Phase 3 | 3000 | HCQ, 400 mg (day-1), then 200 mg for a week (b.i.d.) | Placebo | Use of HCQ as a preventive therapy against COVID-19 | United States | Henry Ford Health System |
| NCT04371744 | AI for QT Interval Analysis of ECG From Smartwatches in Patient Receiving Treatment for Covid-19 (QT-Logs) | Observational, Cohort, Prospective | - | 100 | Not Applicable | - | Measurement of QTc using an AI and ECG data via smartwatches, compare to standard 12 leads ECG | Marseille, France | Assistance Publique Hopitaux De Marseille |
| NCT043329 | Outcomes Related to COVID-19 treated with HCQ Among In-patients with Symptomatic Disease (ORCHID) | Interventional, Randomized | Phase 3 | 510 | HCQ, 400 mg (day-1), then 200 mg for next 5 days (b.i.d.) | Placebo | Determine the COVID Ordinal Scale for patients on day 15 | United States | Massachusetts General Hospital |
| NCT04353245 | Study of Biomarkers in the Long-term Impact of Coronavirus Infection in the Cardiorespiratory System (PostCOVID19) | Observational [Registry], Case-Control | - | 130 | Arm treatment (HCQ + AZM) | - | Fibrosis on cardiac resonance and/or decreased functional capacity on ergo-spirometry | São Paulo, SP, Brazil | University of Sao Paulo General Hospital |
| NCT04372082 | Hydroxychloroquine or Diltiazem-Niclosamide for the Treatment of COVID-19 (HYdILIC) | Interventional, Randomized | Phase 3 | 480 | HCQ, 2200 mg (t.i.d.) during 10 days in addition to SOC; While niclosamide 500 mg × 4 at J1 then 500 mg (b.i.d.) + diltiazem 60 mg (t.i.d.) during 10 days | HCQ, Diltiazem & Niclosamide | Composite criteria- death, clinical worsening, and assisted-ventilation | Lille, France | University Hospital, Lille, France |
| NCT04361422 | Isotretinoin in Treatment of COVID-19 (Randomized) | Interventional, Randomized | Phase 3 | 300 | Isotretinoin, 13-cis retinoic acid 0.5 mg/kg/day b.i.d. for 1 month. Sham compa- HCQ 500 mg/12 h & other drugs | Active Comp: HCQ and other drugs+ isotretinoin | Viral clearance and COVID-19 virus load | Tanta city, Egypt | Tanta University, Egypt |
| NCT04374019 | Novel Agents for Treatment of High-risk COVID-19 Positive Patients | Interventional (Clinical Trial), Randomized | Phase 2 | 240 | HCQ 200 mg (t.i.b.) for 14 days. HCQ combination with AZM, Ivermectin, and Camostat Mesilate are also enrolled | - | Proportion of patients experiencing clinical deterioration | Kentucky, United States | Susanne Arnold, University of Kentucky |
| NCT04382625 | Hydroxychloroquine in SARS-CoV-2 (COVID-19) Pneumonia Trial | Interventional, Randomized (Open Label) | Phase 4 | 120 | HCQ 400 mg × 2 (800 mg) then 200 mg, t.i.b. (600 mg/24 h period) starting 8 h after 1st dose, total 14 doses over 5 days | - | Data collection, Change from Baseline Oxygenation on Day 1-5 | Washington SU, USA | Kootenai Health, United States |
| NCT04333355 | Safety in Convalescent Plasma Transfusion to COVID-19 | Interventional, Open label | Phase 1 | 20 | Convalescent Plasma | - | Adverse effects of administration of convalescent plasma | Mexico | Hospital San Jose Tec de Monterrey, Mexico |
| NCT04358068 | Evaluating the Efficacy of Hydroxychloroquine and Azithromycin to Prevent Hospitalization or Death in Persons With COVID-19 | Interventional, Randomized | Phase 2 | 2000 | HCQ (200 × 2 mg day-1, then 200 mg × 2 for 6 days) + AZM (250 mg × 2 mg- Day 0, then 250 mg once daily for 4 doses (4 days) | Placebo | Proportion of patients’ mortality with COVID-19 | San Diego, United States | National Institute of Allergy and Infectious Diseases (NIAID), USA |
| NCT04373044 | Antiviral Therapy and Baricitinib for the Treatment of Patients with Moderate or Severe COVID-19 | Interventional (Clinical Trial), Open label | Phase 2 | 59 | 1) HCQ, PO t.i.d., 2) lopinavir/ritonavir PO b.i.d., or 3) remdesivir. | - | Proportion of patients requiring invasive mechanical ventilation or dying | United States | University of Southern California, United States |
| NCT04349410 | The Fleming [FMTVDM] Directed CoVid-19 Treatment Protocol (FMTVDM) | Interventional, Randomized | Phase 2, Phase 3 | 500 | HCQ, 200 mg po q 8 hrs (600 mg qD) for 10-days, & HCQ regime with other drugs | - | Improvement in FMTVDM Analyzed by nuclear imaging | United States | The Camelot Foundation, USA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalra, R.S.; Tomar, D.; Meena, A.S.; Kandimalla, R. SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens 2020, 9, 546. https://doi.org/10.3390/pathogens9070546
Kalra RS, Tomar D, Meena AS, Kandimalla R. SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens. 2020; 9(7):546. https://doi.org/10.3390/pathogens9070546
Chicago/Turabian StyleKalra, Rajkumar Singh, Dhanendra Tomar, Avtar Singh Meena, and Ramesh Kandimalla. 2020. "SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings" Pathogens 9, no. 7: 546. https://doi.org/10.3390/pathogens9070546
APA StyleKalra, R. S., Tomar, D., Meena, A. S., & Kandimalla, R. (2020). SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens, 9(7), 546. https://doi.org/10.3390/pathogens9070546