
Comparative Genomics Applied to Systematically Assess Pathogenicity Potential in Shiga Toxin-Producing Escherichia coli O145:H28


Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Typing and Comparative Genomic Analyses
2.2. Identification of Virulence Genes
2.3. Detection of GIs and PAIs
2.4. Statistical Analyses
3. Results
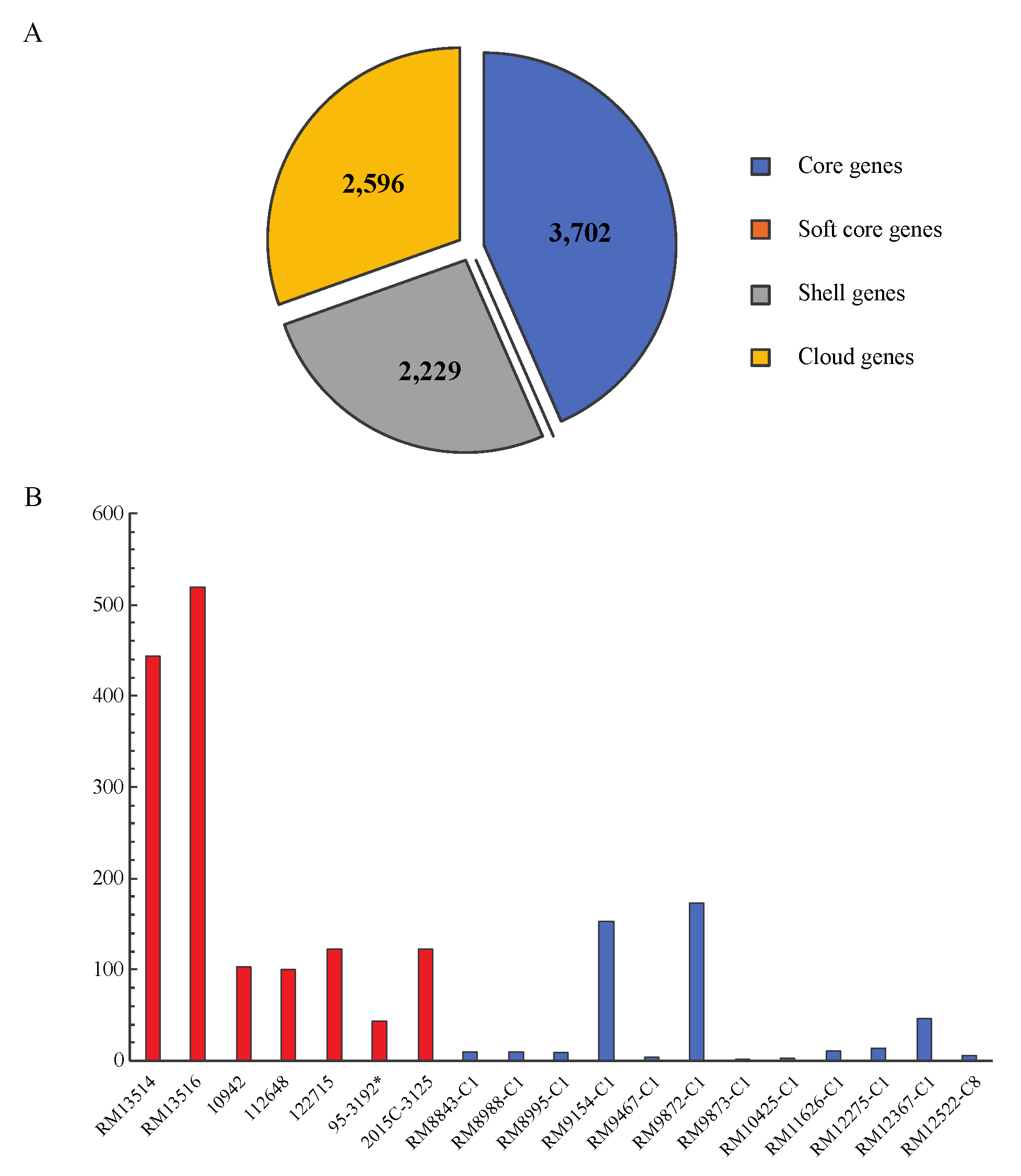
3.1. Comparative Genomics of STEC O145:H28
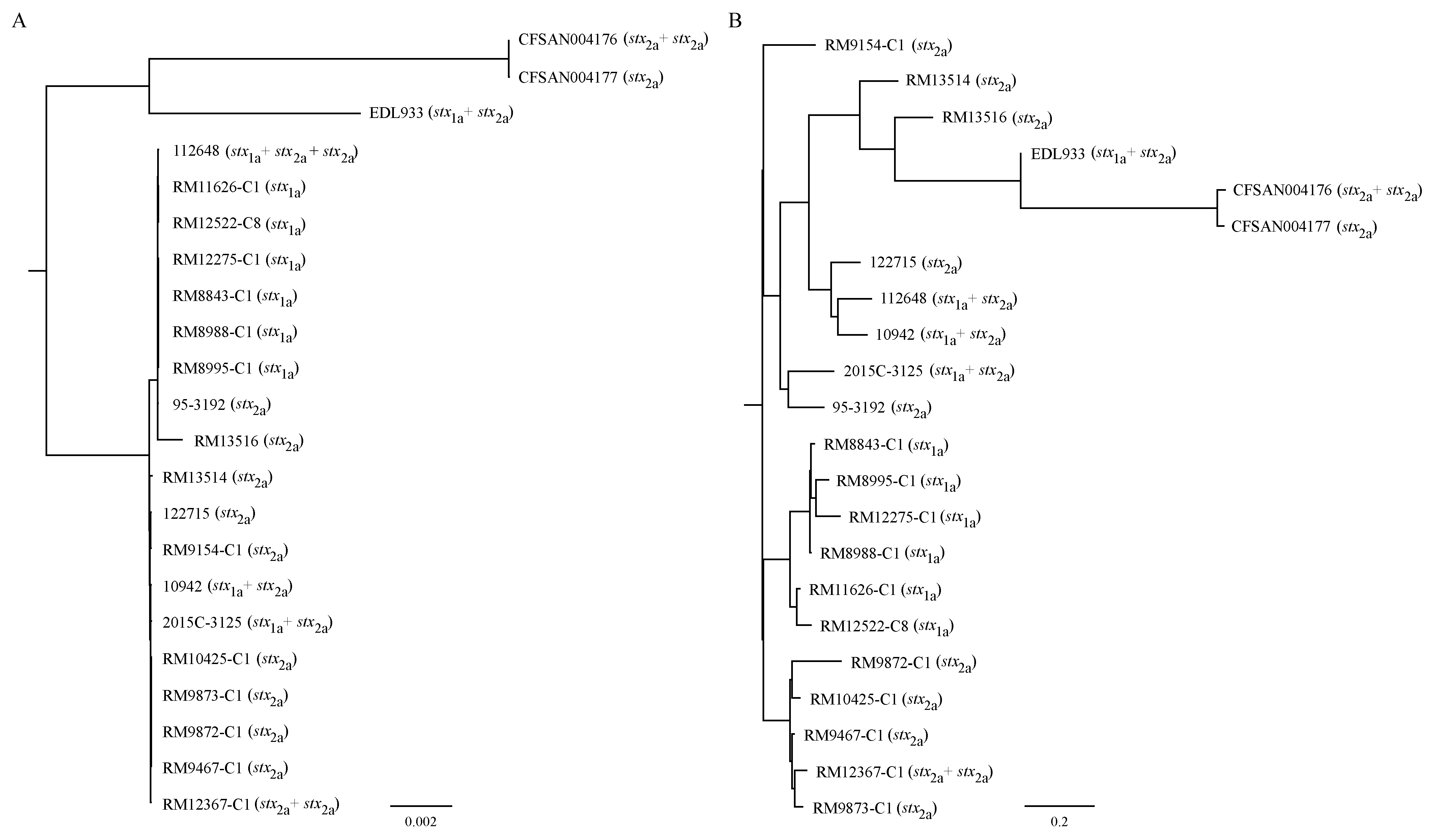
3.2. Strain Relatedness
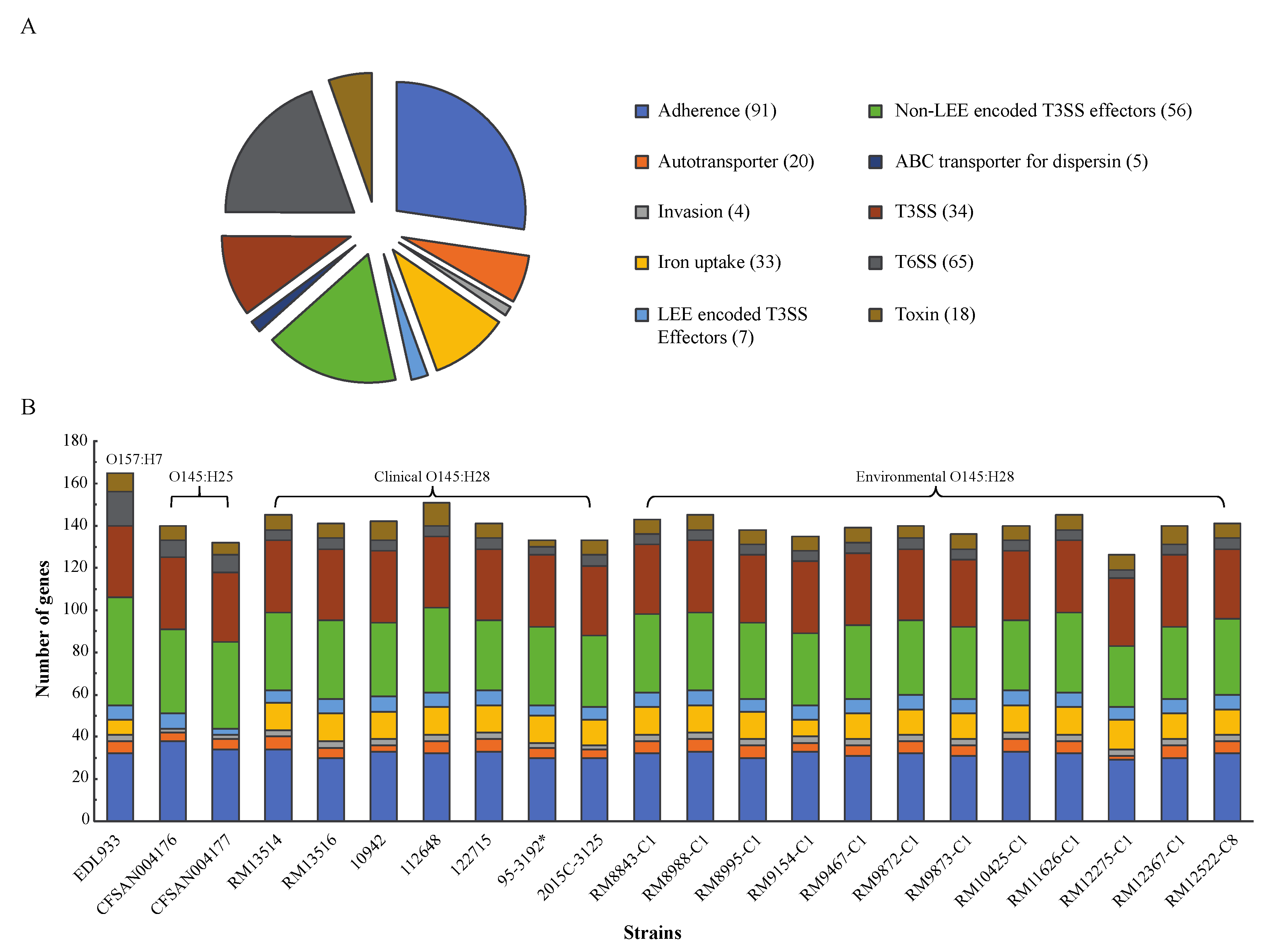
3.3. Conservation of E. coli Virulence Genes in Environmental Strains
3.4. Diversification of Pathogenicity Islands (PAIs) in STEC
3.4.1. LEE
3.4.2. OI-122
3.4.3. OI-57
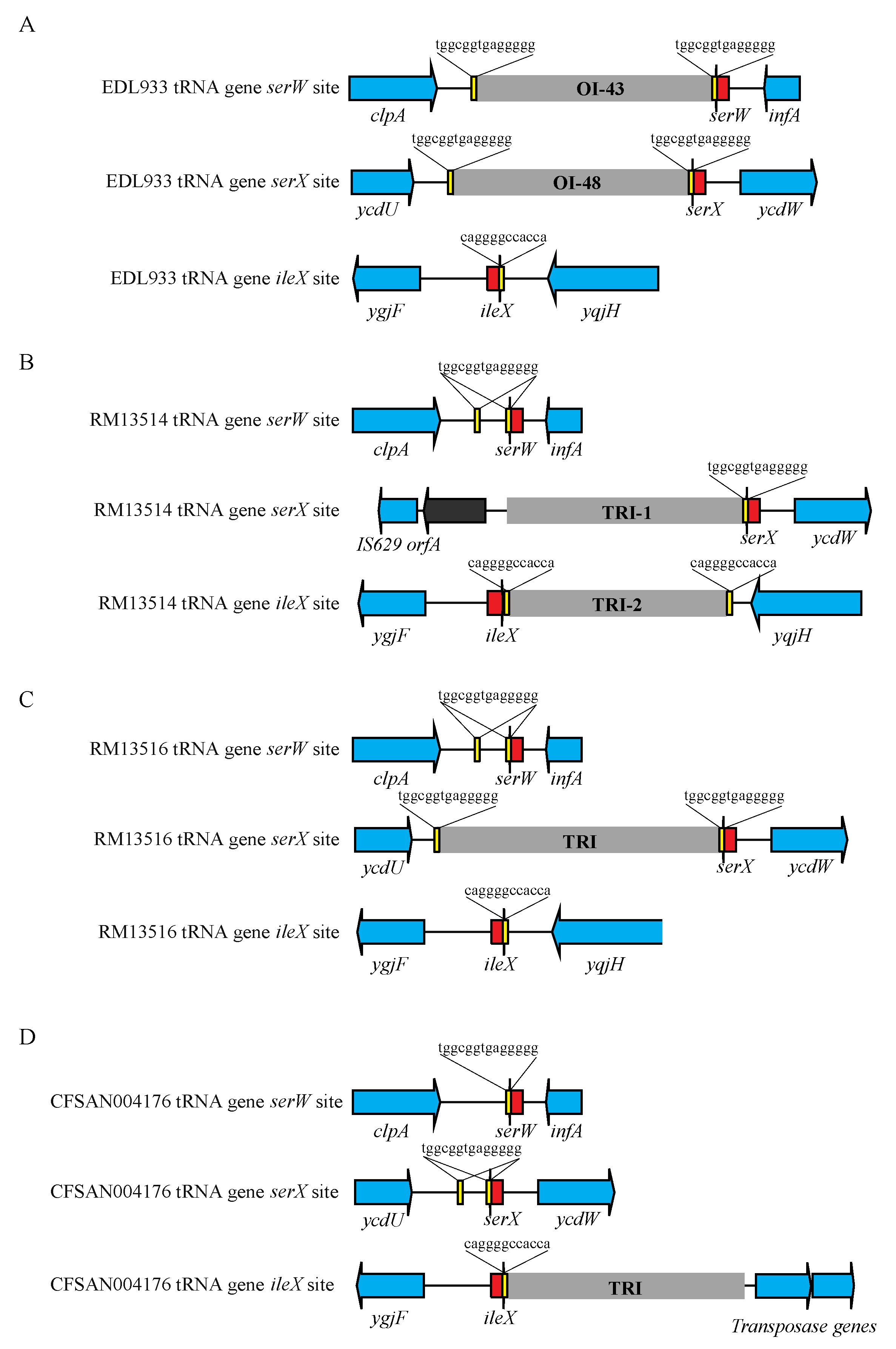
3.4.4. TRI
3.4.5. LAA
3.4.6. Others
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Coombes, B.K.; Gilmour, M.W.; Goodman, C.D. The evolution of virulence in non-O157 Shiga toxin-producing Escherichia coli. Front. Microbiol. 2011, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, R.R.; Henderson, I.R. The evolution of the Escherichia coli phylogeny. Infect. Genet. Evol. 2012, 12, 214–226. [Google Scholar] [CrossRef]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [Green Version]
- Touchon, M.; Hoede, C.; Tenaillon, O.; Barbe, V.; Baeriswyl, S.; Bidet, P.; Bingen, E.; Bonacorsi, S.; Bouchier, C.; Bouvet, O.; et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009, 5, e1000344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [Green Version]
- Cooper, K.K.; Mandrell, R.E.; Louie, J.W.; Korlach, J.; Clark, T.A.; Parker, C.T.; Huynh, S.; Chain, P.S.; Ahmed, S.; Carter, M.Q. Comparative genomics of enterohemorrhagic Escherichia coli O145: H28 demonstrates a common evolutionary lineage with Escherichia coli O157: H7. BMC Genom. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Ooka, T.; Iguchi, A.; Toh, H.; Asadulghani, M.; Oshima, K.; Kodama, T.; Abe, H.; Nakayama, K.; Kurokawa, K.; et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 17939–17944. [Google Scholar] [CrossRef] [Green Version]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Kock, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Brockhurst, M.A.; Harrison, E.; Hall, J.P.J.; Richards, T.; McNally, A.; MacLean, C. The Ecology and Evolution of Pangenomes. Curr. Biol. 2019, 29, R1094–R1103. [Google Scholar] [CrossRef] [PubMed]
- Lupolova, N.; Dallman, T.J.; Matthews, L.; Bono, J.L.; Gally, D.L. Support vector machine applied to predict the zoonotic potential of E. coli O157 cattle isolates. Proc. Natl. Acad. Sci. USA 2016, 113, 11312–11317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, S.A.; Menge, C.; Eichhorn, I.; Semmler, T.; Wieler, L.H.; Pickard, D.; Belka, A.; Berens, C.; Geue, L. The accessory genome of Shiga toxin-producing Escherichia coli defines a persistent colonization type in cattle. Appl. Environ. Microbiol. 2016, 82, 5455–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimizu, Y.; Kirino, Y.; Sato, M.P.; Uno, K.; Sato, T.; Gotoh, Y.; Auvray, F.; Brugere, H.; Oswald, E.; Mainil, J.G.; et al. Large-scale genome analysis of bovine commensal Escherichia coli reveals that bovine-adapted E. coli lineages are serving as evolutionary sources of the emergence of human intestinal pathogenic strains. Genome Res. 2019, 29, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.Q. Decoding the Ecological Function of Accessory Genome. Trends Microbiol. 2017, 25, 6–8. [Google Scholar] [CrossRef]
- Grad, Y.H.; Godfrey, P.; Cerquiera, G.C.; Mariani-Kurkdjian, P.; Gouali, M.; Bingen, E.; Shea, T.P.; Haas, B.J.; Griggs, A.; Young, S.; et al. Comparative genomics of recent Shiga toxin-producing Escherichia coli O104:H4: Short-term evolution of an emerging pathogen. mBio 2013, 4, e00452-12. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Plunkett, G., 3rd; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Mates, A.K.; Sayed, A.K.; Foster, J.W. Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J. Bacteriol. 2007, 189, 2759–2768. [Google Scholar] [CrossRef] [Green Version]
- Montero, D.A.; Velasco, J.; Del Canto, F.; Puente, J.L.; Padola, N.L.; Rasko, D.A.; Farfan, M.; Salazar, J.C.; Vidal, R. Locus of Adhesion and Autoaggregation (LAA), a pathogenicity island present in emerging Shiga Toxin-producing Escherichia coli strains. Sci. Rep. 2017, 7, 7011. [Google Scholar] [CrossRef] [Green Version]
- Mercer, R.; Nguyen, O.; Ou, Q.; McMullen, L.; Ganzle, M.G. Functional Analysis of Genes Comprising the Locus of Heat Resistance in Escherichia coli. Appl. Environ. Microbiol. 2017, 83, e01400-17. [Google Scholar] [CrossRef] [Green Version]
- Juhas, M.; Van Der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, D.A.; Canto, F.D.; Velasco, J.; Colello, R.; Padola, N.L.; Salazar, J.C.; Martin, C.S.; Onate, A.; Blanco, J.; Rasko, D.A.; et al. Cumulative acquisition of pathogenicity islands has shaped virulence potential and contributed to the emergence of LEE-negative Shiga toxin-producing Escherichia coli strains. Emerg. Microbes Infect. 2019, 8, 486–502. [Google Scholar] [CrossRef] [Green Version]
- Bielaszewska, M.; Dobrindt, U.; Gartner, J.; Gallitz, I.; Hacker, J.; Karch, H.; Muller, D.; Schubert, S.; Alexander Schmidt, M.; Sorsa, L.J.; et al. Aspects of genome plasticity in pathogenic Escherichia coli. Int. J. Med. Microbiol. 2007, 297, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.T.; Sowers, E.G.; Wells, J.G.; Greene, K.D.; Griffin, P.M.; Hoekstra, R.M.; Strockbine, N.A. Non-O157 Shiga toxin-producing Escherichia coli infections in the United States, 1983-2002. J. Infect. Dis. 2005, 192, 1422–1429. [Google Scholar] [CrossRef] [Green Version]
- Gould, L.H.; Mody, R.K.; Ong, K.L.; Clogher, P.; Cronquist, A.B.; Garman, K.N.; Lathrop, S.; Medus, C.; Spina, N.L.; Webb, T.H.; et al. Increased recognition of non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000–2010: Epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog. Dis. 2013, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Luna-Gierke, R.E.; Griffin, P.M.; Gould, L.H.; Herman, K.; Bopp, C.A.; Strockbine, N.; Mody, R.K. Outbreaks of non-O157 Shiga toxin-producing Escherichia coli infection: USA. Epidemiol. Infect. 2014, 142, 2270–2280. [Google Scholar] [CrossRef]
- Lorenz, S.C.; Gonzalez-Escalona, N.; Kotewicz, M.L.; Fischer, M.; Kase, J.A. Genome sequencing and comparative genomics of enterohemorrhagic Escherichia coli O145:H25 and O145:H28 reveal distinct evolutionary paths and marked variations in traits associated with virulence & colonization. BMC Microbiol. 2017, 17, 183. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.Q.; Quinones, B.; He, X.; Zhong, W.; Louie, J.W.; Lee, B.G.; Yambao, J.C.; Mandrell, R.E.; Cooley, M.B. An environmental Shiga toxin-producing Escherichia coli O145 clonal population exhibits high-level phenotypic variation that includes virulence traits. Appl. Environ. Microbiol. 2016, 82, 1090–1101. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.Q.; Pham, A.; Du, W.X.; He, X. Differential induction of Shiga toxin in environmental Escherichia coli O145:H28 strains carrying the same genotype as the outbreak strains. Int. J. Food Microbiol. 2021, 339, 109029. [Google Scholar] [CrossRef]
- Mercer, R.G.; Zheng, J.; Garcia-Hernandez, R.; Ruan, L.; Ganzle, M.G.; McMullen, L.M. Genetic determinants of heat resistance in Escherichia coli. Front. Microbiol. 2015, 6, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Gordon, D.; Denamur, E. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology (Reading) 2015, 161, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Tetzschner, A.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and Easy In Silico Serotyping of Escherichia coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Burland, V.; Shao, Y.; Perna, N.T.; Plunkett, G.; Sofia, H.J.; Blattner, F.R. The complete DNA sequence and analysis of the large virulence plasmid of Escherichia coli O157:H7. Nucleic Acids Res. 1998, 26, 4196–4204. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Murase, K.; Sato, M.P.; Toyoda, A.; Itoh, T.; Mainil, J.G.; Piérard, D.; Yoshino, S.; Kimata, K.; Isobe, J. Differential dynamics and impacts of prophages and plasmids on the pangenome and virulence factor repertoires of Shiga toxin-producing Escherichia coli O145: H28. Microbial. Genom. 2020, 6, e000323. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.N.; Lindsey, R.L.; Garcia-Toledo, L.; Rowe, L.A.; Batra, D.; Whitley, S.W.; Drapeau, D.; Stoneburg, D.; Martin, H.; Juieng, P.; et al. High-Quality Whole-Genome Sequences for 77 Shiga Toxin-Producing Escherichia coli Strains Generated with PacBio Sequencing. Genome Announc. 2018, 6, e00391-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, B.J. Escherichia coli and Salmonella Typhimurium: Cellular and Molecular Biology; Neidhardt, F.C., Ed.; ASM Press: Washington, DC, USA, 1996. [Google Scholar]
- Iguchi, A.; Iyoda, S.; Kikuchi, T.; Ogura, Y.; Katsura, K.; Ohnishi, M.; Hayashi, T.; Thomson, N.R. A complete view of the genetic diversity of the Escherichia coli O-antigen biosynthesis gene cluster. DNA Res. 2015, 22, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Rothemund, D.; Curd, H.; Reeves, P.R. Species-wide variation in the Escherichia coli flagellin (H-antigen) gene. J. Bacteriol. 2003, 185, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Schmidt, H.; Zhang, W.L.; Hemmrich, U.; Jelacic, S.; Brunder, W.; Tarr, P.I.; Dobrindt, U.; Hacker, J.; Karch, H. Identification and characterization of a novel genomic island integrated at selC in locus of enterocyte effacement-negative, Shiga toxin-producing Escherichia coli. Infect. Immun. 2001, 69, 6863–6873. [Google Scholar] [CrossRef] [Green Version]
- Schubert, S.; Rakin, A.; Karch, H.; Carniel, E.; Heesemann, J. Prevalence of the “high-pathogenicity island” of Yersinia species among Escherichia coli strains that are pathogenic to humans. Infect. Immun. 1998, 66, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Michelacci, V.; Tozzoli, R.; Caprioli, A.; Martinez, R.; Scheutz, F.; Grande, L.; Sanchez, S.; Morabito, S. A new pathogenicity island carrying an allelic variant of the Subtilase cytotoxin is common among Shiga toxin producing Escherichia coli of human and ovine origin. Clin. Microbiol. Infect. 2013, 19, E149–E156. [Google Scholar] [CrossRef] [Green Version]
- Wells, T.J.; Sherlock, O.; Rivas, L.; Mahajan, A.; Beatson, S.A.; Torpdahl, M.; Webb, R.I.; Allsopp, L.P.; Gobius, K.S.; Gally, D.L.; et al. EhaA is a novel autotransporter protein of enterohemorrhagic Escherichia coli O157:H7 that contributes to adhesion and biofilm formation. Environ. Microbiol. 2008, 10, 589–604. [Google Scholar] [CrossRef]
- Wells, T.J.; McNeilly, T.N.; Totsika, M.; Mahajan, A.; Gally, D.L.; Schembri, M.A. The Escherichia coli O157:H7 EhaB autotransporter protein binds to laminin and collagen I and induces a serum IgA response in O157:H7 challenged cattle. Environ. Microbiol. 2009, 11, 1803–1814. [Google Scholar] [CrossRef]
- Journet, L.; Cascales, E. The Type VI Secretion System in Escherichia coli and Related Species. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamovic, L.; Tozzoli, R.; Michelacci, V.; Minelli, F.; Marziano, M.L.; Caprioli, A.; Morabito, S. OI-57, a genomic island of Escherichia coli O157, is present in other seropathotypes of Shiga toxin-producing E. coli associated with severe human disease. Infect. Immun. 2010, 78, 4697–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielaszewska, M.; Middendorf, B.; Tarr, P.I.; Zhang, W.; Prager, R.; Aldick, T.; Dobrindt, U.; Karch, H.; Mellmann, A. Chromosomal instability in enterohaemorrhagic Escherichia coli O157:H7: Impact on adherence, tellurite resistance and colony phenotype. Mol. Microbiol. 2011, 79, 1024–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney, J.O.; McNally, A.; O’Connell, M.J. Why prokaryotes have pangenomes. Nat. Microbiol. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Rohmer, L.; Hocquet, D.; Miller, S.I. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011, 19, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Hottes, A.K.; Freddolino, P.L.; Khare, A.; Donnell, Z.N.; Liu, J.C.; Tavazoie, S. Bacterial adaptation through loss of function. PLoS Genet. 2013, 9, e1003617. [Google Scholar] [CrossRef]
- Carter, M.Q.; Parker, C.T.; Louie, J.W.; Huynh, S.; Fagerquist, C.K.; Mandrell, R.E. RcsB contributes to the distinct stress fitness among Escherichia coli O157:H7 curli variants of the 1993 hamburger-associated outbreak strains. Appl. Environ. Microbiol. 2012, 78, 7706–7719. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.Q.; Louie, J.W.; Huynh, S.; Parker, C.T. Natural rpoS mutations contribute to population heterogeneity in Escherichia coli O157:H7 strains linked to the 2006 US spinach-associated outbreak. Food Microbiol. 2014, 44, 108–118. [Google Scholar] [CrossRef]
- Weissman, S.J.; Beskhlebnaya, V.; Chesnokova, V.; Chattopadhyay, S.; Stamm, W.E.; Hooton, T.M.; Sokurenko, E.V. Differential stability and trade-off effects of pathoadaptive mutations in the Escherichia coli FimH adhesin. Infect. Immun. 2007, 75, 3548–3555. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Brauner, A.; Li, Y.; Normark, S. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J. Infect. Dis. 2000, 181, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Kai-Larsen, Y.; Luthje, P.; Chromek, M.; Peters, V.; Wang, X.; Holm, A.; Kadas, L.; Hedlund, K.O.; Johansson, J.; Chapman, M.R.; et al. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. 2010, 6, e1001010. [Google Scholar] [CrossRef] [PubMed]
- Tukel, C.; Raffatellu, M.; Humphries, A.D.; Wilson, R.P.; Andrews-Polymenis, H.L.; Gull, T.; Figueiredo, J.F.; Wong, M.H.; Michelsen, K.S.; Akcelik, M.; et al. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol. Microbiol. 2005, 58, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.Q.; Brandl, M.T.; Kudva, I.T.; Katani, R.; Moreau, M.R.; Kapur, V. Conditional Function of Autoaggregative Protein Cah and Common cah Mutations in Shiga Toxin-Producing Escherichia coli. Appl. Environ. Microbiol. 2018, 84, e01739-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanassi, D.G.; Suh, G.S.; Nikaido, H. Role of outer membrane barrier in efflux-mediated tetracycline resistance of Escherichia coli. J. Bacteriol. 1995, 177, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, A.; Alfano, J.R. Disabling surveillance: Bacterial type III secretion system effectors that suppress innate immunity. Cell. Microbiol. 2004, 6, 1027–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Mei, M.; Yu, C.; Shen, W.; Ma, L.; He, J.; Yi, L. The Functions of Effector Proteins in Yersinia Virulence. Pol. J. Microbiol. 2016, 65, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Goosney, D.L.; DeVinney, R.; Pfuetzner, R.A.; Frey, E.A.; Strynadka, N.C.; Finlay, B.B. Enteropathogenic E. coli translocated intimin receptor, Tir, interacts directly with alpha-actinin. Curr. Biol. 2000, 10, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Petersen, L.; Bollback, J.P.; Dimmic, M.; Hubisz, M.; Nielsen, R. Genes under positive selection in Escherichia coli. Genome Res. 2007, 17, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Tchesnokova, V.; McVeigh, A.; Kisiela, D.I.; Dori, K.; Navarro, A.; Sokurenko, E.V.; Savarino, S.J. Adaptive evolution of class 5 fimbrial genes in enterotoxigenic Escherichia coli and its functional consequences. J. Biol. Chem. 2012, 287, 6150–6158. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.K.; Levine, M.M.; Morison, J.; Phillips, A.; Barry, E.M. CfaE tip mutations in enterotoxigenic Escherichia coli CFA/I fimbriae define critical human intestinal binding sites. Cell. Microbiol. 2009, 11, 742–754. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | a Sources (Location, Year) | b Serotype | Phylogroup/Genotype | stx Genes | Chromosome (bp)/ GenBank Accession # | c Plasmids (bp)/GenBank Accession | References | |
|---|---|---|---|---|---|---|---|---|
| pEHEC | Others | |||||||
| MG1655 | Stool (USA, 1922) | O16:H48 | A/ST10 | N/A | 4,641,652/U00096.3 | N/A | N/A | [39] |
| EDL933 | Ground beef (USA, 1982) | O157:H7 | E/ST11 | stx1a + stx2a | 5,528,445/AE005174.2 | 92,077/AF074613.1 | N/A | [17,40] |
| CFSAN004176 | Clinical (USA, 2003) | O145:H25 | B1/ST5309 | stx2a + stx2a | 5,193,734/CP014583.1 | 52,297/CP012493.1 | 95,721/CP012491.1; 34,714/CP012492.1 | [28] |
| CFSAN004177 | Clinical (USA, 2004) | O145:H25 | B1/ST5309 | stx2a | 5,191,331/CP014670.1 | 52,297/CP012495.1 | 96,228/CP012494.1; 34,714/CP012496.1 | [28] |
| RM13514 | Clinical (USA, 2010) | O145:H28 | E/ST32 | stx2a | 5,585,613/CP006027.1 | 87,120/CP006028.1 | 64,561/CP006029.1 | [6] |
| RM13516 | Clinical (Belgium, 2007) | O145:H28 | E/ST6130 | stx2a | 5,402,276/CP006262.1 | 98,066/CP006263.1 | 58,666/CP006264.1 | [6] |
| 10942 | Clinical (Japan, 2011) | O145:H28 | E/ST32 | stx1a + stx2a | 5,374,674/AP019703.1 | 92,337/AP019704.1 | 71,161/AP019705.1 | [41] |
| 112648 | Clinical (Japan, 2011) | O145:H28 | E/ST32 | stx1a + stx2a + stx2a | 5,488,534/AP019706.1 | 91,036/AP019707.1 | N/A | [41] |
| 122715 | Clinical (Japan, 2012) | O145:H28 | E/ST32 | stx2a | 5,418,961/AP019708.1 | 86,874/AP019709.1 | 48,572/AP019710.1 | [41] |
| 95-3192 | Clinical (USA, NA) | O145:H28 | E/ST32 | stx2a | 5,385,516/CP027362.1 | N/A | N/A | [42] |
| 2015C-3125 | Clinical (USA, 2014) | O145:H28 | E/ST32 | stx1a + stx2a | 5,471,132/CP027763.1 | 66,944/CP027764.1 | 66,388/CP027765.1 | [42] |
| RM8843-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx1a | 5,458,415/CP035772.1 | 88,752/CP035773.1 | N/A | [30] |
| RM8988-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx1a | 5,458,186/CP035770.1 | 88,752/CP035771.1 | N/A | [30] |
| RM8995-C1 | Sediment (USA, 2009) | O145:H28 | E/ST32 | stx1a | 5,457,980/CP031355.1 | 88,747/CP031354.1 | N/A | [30] |
| RM9154-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx2a | 5,205,721/CP031353.1 | 86,711/CP031352.1 | 187,274/CP031350.1; 96,355/CP031351.1 | [30] |
| RM9467-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx2a | 5,385,895/CP031349.1 | 89,518/CP031348.1 | N/A | [30] |
| RM9872-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx2a | 5,385,904/CP024659.1 | 89,518/CP024660.1 | N/A | [30] |
| RM9873-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx2a | 5,385,819/CP031347.1 | 89,515/CP031346.1 | N/A | [30] |
| RM10425-C1 | Cattle (USA, 2009) | O145:H28 | E/ST32 | stx2a | 5,343,037/CP031343.1 | 89,519/CP031342.1 | N/A | [30] |
| RM11626-C1 | Cattle (USA, 2010) | O145:H28 | E/ST32 | stx1a | 5,419,044/CP035768.1 | 86,875/CP035769.1 | N/A | [30] |
| RM12275-C1 | Cattle (USA, 2010) | O145:H28 | E/ST32 | stx1a | 5,408,598/CP031341.1 | 88,744/CP031340.1 | N/A | [30] |
| RM12367-C1 | Water (USA, 2010) | O145:H28 | E/ST32 | stx2a + stx2a | 5,472,396/CP031345.1 | 90,831/CP031344.1 | N/A | [30] |
| RM12522-C8 | Cattle (USA, 2010) | O145:H28 | E/ST32 | stx1a | 5,418,923/CP035767.1 | 86,874/CP035766.1 | N/A | [30] |
| PAIs/GIs | Length (bp) /%GC | Sources of Query Sequences | GenBank Accession #/Positions | References |
|---|---|---|---|---|
| Locus of Enterocyte Effacement (LEE) | 43,418/40.9 | STEC O157:H7 str. EDL933 | AE005174.2/4,649,862–4,693,279 | [17] |
| Pathogenicity Island OI-122 | 23,455/46.3 | STEC O157:H7 str. EDL933 | AE005174.2/3,919,348–3,942,802 | [17] |
| Pathogenicity Island OI-57 | 80,502/51.4 | STEC O157:H7 str. EDL933 | AE005174.2/1,849,324–1,929,825 | [17] |
| Tellurite Resistance Island (TRI) | 87,548/48.0 | STEC O157:H7 str. EDL933 | AE005174.2/1,454,242–1,541,789 | [17] |
| Locus of Adhesion and Autoaggregation (LAA) | 86,353/48.6 | STEC O91:H21 str. B2F1 | AFDQ01000026.1/385,984–472,336 | [20] |
| Locus of Proteolysis Activity (LPA) | 37,710/47.4 | STEC O91:H- str. 4797/97 | AJ278144.1/1–37,710 | [47] |
| High-Pathogenicity Island (HPI) | 36,448/56.4 | Yersinia pestis | AL031866.1/78,113–114,560 | [48] |
| Subtilase Encoding Pathogenicity Island (SE-PAI) | 8058/46.2 | E. coli str. ED32 | JQ994271.1/1–8058 | [49] |
| Acid Fitness Island (AFI) | 13,620/46.0 | E. coli str. MG1655 | U00096.3/3,653,961–3,667,580 | [19] |
| Locus of Heat Resistance (LHR) | 14,981/62.2 | E. coli str. P12b | CP002291.1/319,821–304,841 | [21] |
| Strains | a LEE | b OI-122 | c OI-57 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Integration Site | Chromosomal Location | Size (bp)/CDS | Integration Site | Chromosomal Location | Size (bp)/CDS | Integration Site | Chromosomal Location | Size (bp)/CDS | |
| EDL933 | selC | 4,649,862–4,693,279 | 43,418/52 | pheV/pheU | 3,919,348–3,942,802 | 23,455/27 | yciD (ompW) | 1,849,324–1,929,825 | 80,502/107 |
| CFSAN004176 | pheV | 2,363,880–2,425,764 | 61,885/60 | pheV/pheU | 3,818,649–3,780,408 | 38,242/40 | NA | NA | NA |
| CFSAN004177 | pheV | 2,825,461–2,763,585 | 61,877/61 | pheV/pheU | 1,370,795–1,409,031 | 38,237/40 | NA | NA | NA |
| RM13514 | selC | 4,558,936–4,605,536 | 46,601/55 | pheV/pheU | 5,269,766–5,222,118 | 47,649/47 | ompW | 1,591,159–1,633,674 | 42,516/51 |
| RM13516 | selC | 4,410,578–4,458,362 | 47,785/57 | pheV/pheU | 5,188,896–5,242,157 | 53,262/53 | ompW | 1,569,959–1,615,548 | 45,590/55 |
| 10942 | selC | 4,428,998–4,475,832 | 46,835/56 | pheV/pheU | 5,060,248–5,047,349 | 12,900/10 | ompW | 1,553,610–1,597,236 | 43,627/50 |
| 112648 | selC | 4,538,738–4,585,714 | 46,977/56 | pheV/pheU | 5,203,887–5,157,474 | 46,414/37 | ompW | 1,631,815–1,715,209 | 83,395/102 |
| 122715 | selC | 4,436,958–4,483,934 | 46,977/56 | pheV/pheU | 5,103,184–5,055,469 | 47,716/39 | ompW | 1,575,808–1,619,718 | 43,911/50 |
| 95-3192 | selC | 2,903,392–2,950,270 | 46,879/55 | pheV/pheU | 3,567,963–3,521,794 | 46,170/33 | ompW | 506,141–491,757 | 14,385/24 |
| 2015C-3125 | selC | 4,369,007–4,416,023 | 47,017/55 | pheV/pheU | 5,029,590–4,987,764 | 41,827/34 | ompW | 1,444,864–1,459,247 | 14,384/24 |
| RM8843-C1 | selC | 4,473,390–4,520,271 | 46,882/56 | pheV/pheU | 5,142,570–5,091,970 | 50,601/55 | ompW | 2,204,394–2,248,382 | 43,989/59 |
| RM8988-C1 | selC | 4,473,157–4,520,039 | 46,883/56 | pheV/pheU | 5,141,855–5,091,255 | 50,601/55 | ompW | 1,628,125–1,613,741 | 14,385/26 |
| RM8995-C1 | selC | 4,472,986–4,519,862 | 46,877/56 | pheV/pheU | 5,142,141–5,091,544 | 50,598/55 | ompW | 3,201,502–3,157,514 | 43,989/59 |
| RM9154-C1 | selC | 4,237,174–4,284,056 | 46,883/56 | pheV/pheU | 4,891,189–4,855,660 | 35,530/36 | ompW | 159,456–186,929 | 27,474/44 |
| RM9467-C1 | selC | 4,411,115–4,458,137 | 47,023/56 | pheV/pheU | 5,071,448–5,029,449 | 42,000/42 | ompW | 1,574,704–1,618,656 | 43,953/58 |
| RM9872-C1 | selC | 4,411,123–4,458,145 | 47,023/58 | pheV/pheU | 5,071,457–5,029,079 | 42,379/44 | ompW | 2,099,371–2,084,987 | 14,385/24 |
| RM9873-C1 | selC | 4,411,053–4,458,073 | 47,021/57 | pheV/pheU | 5,071,377–5,029,379 | 41,999/42 | ompW | 1,574,682–1,618,634 | 43,953/58 |
| RM10425-C1 | selC | 4,368,259–4,415,280 | 47,022/56 | pheV/pheU | 5,028,591–4,986,592 | 42,000/42 | ompW | 2,099,365–2,084,981 | 14,385/26 |
| RM11626-C1 | selC | 4,439,648–4,486,530 | 46,883/56 | pheV/pheU | 5,104,582–5,058,414 | 46,169/41 | ompW | 3,172,643–3,158,259 | 14,385/26 |
| RM12275-C1 | selC | 4,472,938–4,519,814 | 46,877/56 | NA | NA | NA | ompW | 1,622,111–1,666,070 | 43,960/59 |
| RM12367-C1 | selC | 4,497,622–4,544,644 | 47,023/56 | pheV/pheU | 5,157,953–5,115,955 | 41,999/42 | ompW | 1,575,949–1,713,896 | 137,948/191 |
| RM12522-C8 | selC | 4,439,541–4,486,422 | 46,882/56 | pheV/pheU | 5,104,468–5,058,301 | 46,168/41 | ompW | 1,625,877–1,652,995 | 27,119/40 |
| Strains | a Tellurite Resistance Island (TRI) | Locus of Adherence and Autoaggregation (LAA) Module Location/Size (bp) | Acid Fitness Island (AFI) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Related tRNA Genes | Chromosomal Locations | Size (bp)/ CDS | I | II | III | IV | Chromosomal Locations | Size(bp)/ CDS | |
| EDL933 | OI-43/serW; OI-48/serX | OI-43:1,058,620–1,146,182 | 87,563/106 | OI-43/864 | OI-43/ 5777 | OI-43/ 11,256 | OI-43/15,455 | 4,454,268–4,476,943 | 22,676/21 |
| OI-48:1,454,242–1,541,789 | 87,548/105 | OI-48/864 | OI-48/ 5777 | OI-48/ 11,255 | OI-48/15,455 | ||||
| CFSAN004176 | ileX | 3,679,064–3,626,942 | 52,123/61 | NA | b TRI/ 4994 | NA | TRI/2486 | 3,171,287–3,157,668 | 13,620/13 |
| CFSAN004176 | ileX | 1,510,368–1,562,482 | 52,115/60 | NA | b TRI/ 4995 | NA | TRI/2486 | 2,018,117–2,031,735 | 13,619/13 |
| RM13514 | TRI-1/serX; TRI-2/ileX | TRI-1:1,241,544–1,322,994 | 81,451/103 | TRI-1/863 | TRI-1/ 5777 b TRI-2/ 4950 | TRI-1/ 9945 | TRI-1/15,593; OI-122/13,972; TRI-2/2486 | 4,360,251–4,382,890 | 22,640/24 |
| TRI-2:3,926,463–3,861,137 | 65,327/67 | ||||||||
| RM13516 | serX | 1,206,887–1,303,036 | 96,150/111 | TRI/863 | TRI/5776 | TRI/7083 | TRI/14,084; OI-122/19,082; c IE06/19,939 | 4,224,613–4,247,252 | 22,640/24 |
| 10942 | serX | 1,202,851–1,284,087 | 81,237/97 | TRI/863 | TRI/5777 | TRI/9945 | TRI/14,142 | 4,243,061–4,265,700 | 22,640/20 |
| 112648 | serX | 1,280,897–1,362,131 | 81,235/97 | TRI/863 | TRI/5777 | TRI/9696 | TRI/14,140; OI-122/13,972 | 4,352,804–4,375,443 | 22,640/20 |
| 122715 | serX | 1,223,578–1,303,501 | 79,924/94 | TRI/863 | TRI/ 5777 | TRI/ 9945 | TRI/14,142; OI-122/15,273 | 4,251,163–4,273,802 | 22,640/20 |
| 95-3192 | serX | 856,722–775,651 | 81,072/86 | TRI/862 | TRI/ 5766 | TRI/ 9944 | TRI/14,141; OI-122/13,960 | 2,701,442–2,724,081 | 22,640/21 |
| 2015C-3125 | serX | 1,095,410–1,175,328 | 79,919/86 | TRI/862 | TRI/ 5775 | TRI/ 9945 | TRI/14,142; OI-122/15,218 | 4,183,005–4,205,643 | 22,639/21 |
| RM8843-C1 | serX | 1,852,148–1,933,384 | 81,237/99 | TRI/863 | TRI/ 5777 | TRI/ 9696 | TRI/14,142; OI-122/17,759 | 4,287,369–4,310,008 | 22,640/22 |
| RM8988-C1 | serX | 1,980,371–1,899,135 | 81,237/99 | TRI/863 | TRI/ 5777 | TRI/ 9696 | TRI/14,142; OI-122/17,759 | 4,287,136–4,309,775 | 22,640/22 |
| RM8995-C1 | serX | 2,961,815–2,880,586 | 81,230/99 | TRI/863 | TRI/ 5777 | TRI/ 9695 | TRI/14,140; OI-122/17,758 | 4,286,971–4,309,610 | 22,640/22 |
| RM9154-C1 | serX | 1,222,582–1,291,597 | 69,016/90 | TRI/863 | TRI/ 5777 | TRI/ 9945 | TRI/14,142; OI-122/3,488 | 4,036,160–4,058,799 | 22,640/22 |
| RM9467-C1 | serX | 1,223,947–1,305,183 | 81,237/99 | TRI/863 | TRI/ 5777 | TRI/ 9946 | TRI/14,141; OI-122/15,224 | 4,225,103–4,247,742 | 22,640/22 |
| RM9872-C1 | serX | 2,450,129–2,368,893 | 81,237/103 | TRI/863 | TRI/ 5777 | TRI/ 9946 | TRI/14,142; OI-122/15,224 | 4,225,111–4,247,750 | 22,640/24 |
| RM9873-C1 | serX | 1,223,939–1,305,171 | 81,233/99 | TRI/863 | TRI/ 5777 | TRI/ 9946 | TRI/14,140; OI-122/15,223 | 4,225,047–4,247,685 | 22,639/22 |
| RM10425-C1 | serX | 2,407,270–2,326,034 | 81,237/99 | TRI/863 | TRI/ 5777 | TRI/ 9946 | TRI/14,142; OI-122/15,224 | 4,182,248–4,204,887 | 22,640/22 |
| RM11626-C1 | serX | 2,879,078–2,796,529 | 82,550/101 | TRI/863 | TRI/ 5777 | TRI/ 9696 | TRI/14,142; OI-122/13,972 | 4,253,636–4,276,275 | 22,640/22 |
| RM12275-C1 | serX | 1,269,895–1,351,122 | 81,228/99 | TRI/863 | TRI/ 5777 | TRI/ 9695 | TRI/14,141 | 4,286,927–4,309,564 | 22,638/22 |
| RM12367-C1 | serX | 1,223,885–1,305,121 | 81,237/99 | TRI/863 | TRI/ 5777 | TRI/ 9946 | TRI/14,142; OI-122/15,224 | 4,311,611–4,334,250 | 22,640/22 |
| RM12522-C8 | serX | 1,274,000–1,356,546 | 82,547/101 | TRI/863 | TRI/ 5777 | TRI/ 9696 | TRI/14,142; OI-122/13,971 | 4,253,530–4,276,169 | 22,640/22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carter, M.Q.; Laniohan, N.; Lo, C.-C.; Chain, P.S.G. Comparative Genomics Applied to Systematically Assess Pathogenicity Potential in Shiga Toxin-Producing Escherichia coli O145:H28. Microorganisms 2022, 10, 866. https://doi.org/10.3390/microorganisms10050866
Carter MQ, Laniohan N, Lo C-C, Chain PSG. Comparative Genomics Applied to Systematically Assess Pathogenicity Potential in Shiga Toxin-Producing Escherichia coli O145:H28. Microorganisms. 2022; 10(5):866. https://doi.org/10.3390/microorganisms10050866
Chicago/Turabian StyleCarter, Michelle Qiu, Nicole Laniohan, Chien-Chi Lo, and Patrick S. G. Chain. 2022. "Comparative Genomics Applied to Systematically Assess Pathogenicity Potential in Shiga Toxin-Producing Escherichia coli O145:H28" Microorganisms 10, no. 5: 866. https://doi.org/10.3390/microorganisms10050866
APA StyleCarter, M. Q., Laniohan, N., Lo, C. -C., & Chain, P. S. G. (2022). Comparative Genomics Applied to Systematically Assess Pathogenicity Potential in Shiga Toxin-Producing Escherichia coli O145:H28. Microorganisms, 10(5), 866. https://doi.org/10.3390/microorganisms10050866