
Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae


Abstract
:1. Introduction
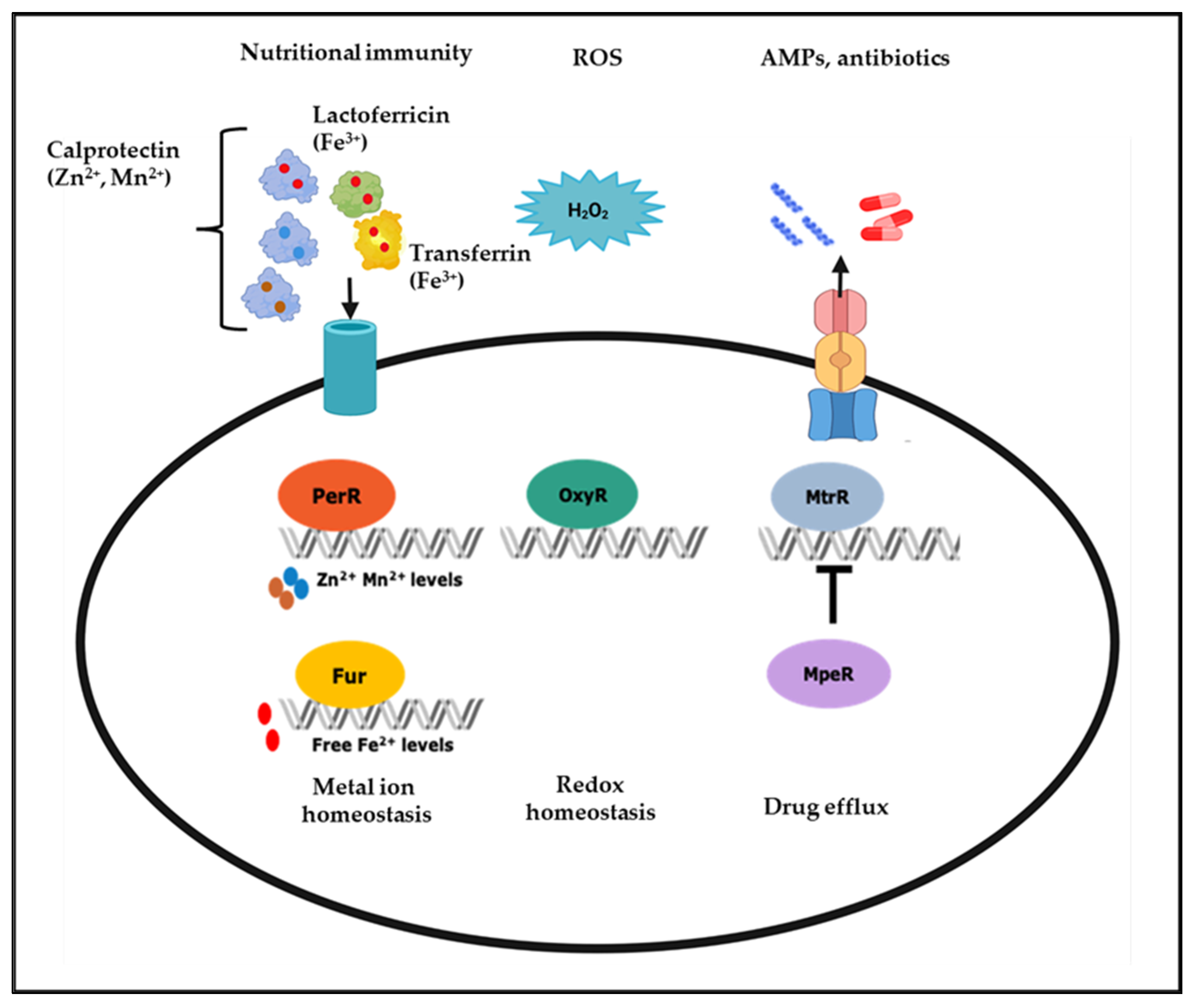
2. DNA-Binding Transcriptional Regulators
3. Phase Variation
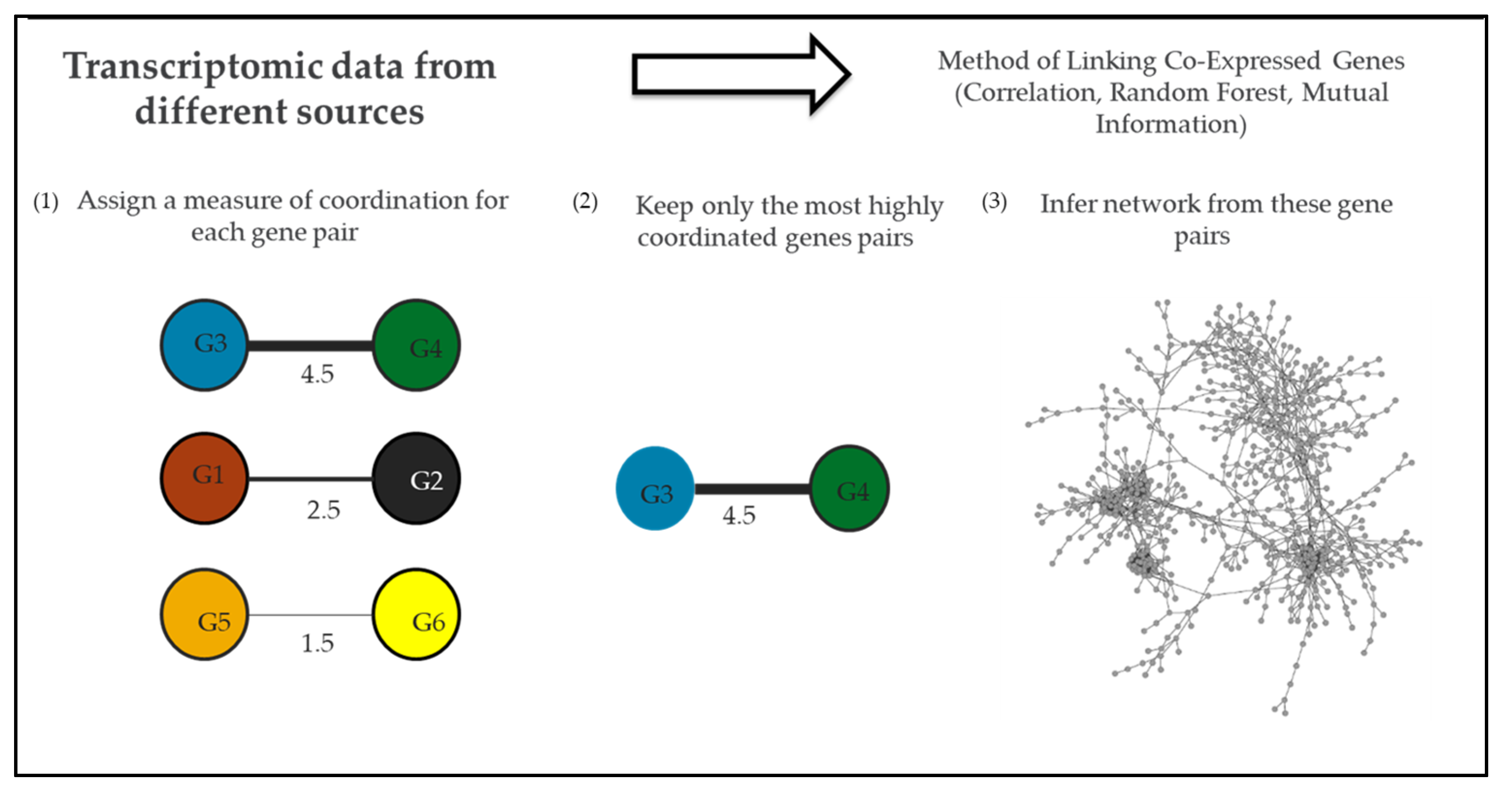
4. Global Gene Co-Expression Network of N. gonorrhoeae
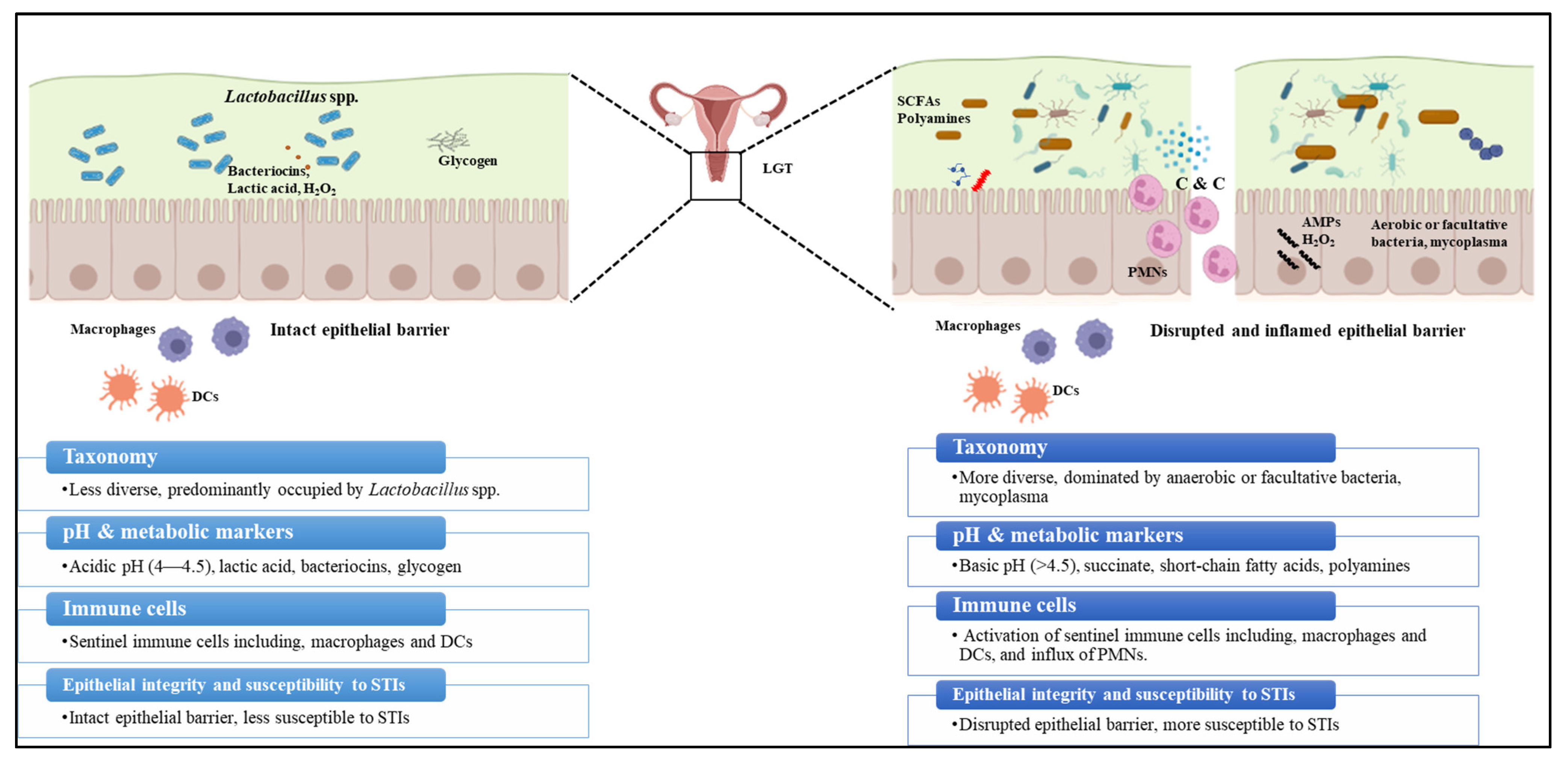
5. Interactions between N. gonorrhoeae and the FGT Microbiome
6. Application of Gene Regulatory Data to Modeling of the Female Genital Tract Microbiome

{kind=link}
{kind=link}
{kind=link}
| Study | Input Data (Features) | Modeling | Major Inferences from the Study (Labels) |
|---|---|---|---|
| [162] | A large longitudinal study looking at more than 3620 women with high Nugent scores | Correlative | There is an association between a high Nugent score and acquisition of N. gonorrhoeae, C. trachomatis, or T. vaginalis infection |
| [163] | 16S amplicon data of vaginal swabs from women from four ethnic/racial groups | Correlative | Prediction of T. vaginalis infection is associated with high bacterial diversity and reduction in Lactobacillus spp. |
| [158] | An analysis of vaginal samples from women who have experienced preterm or term births (control) using 16S amplicons, metagenomic and metatranscriptomic sequencing was carried out | Associative model using a Mann–Whitney U test and assigning weights to these taxa using L1-regularized logistic regression | The abundance of Lactobacillus spp. No difference between pregnant and non-pregnant women Differs in preterm and full-term pregnancies Prediction of preterm birth based on selecting OTUs associated with premature birth Premature birth is significantly associated with four taxa: Sneathia amnii, BV-associated bacterium 1 (BVAB1), Prevotella cluster 2, and TM7-H1 |
| [164] | 16S amplicon timescale data of vaginal samples collected for each subject across 16 weeks | Vagina-specific dynamic microbial interaction network (MIN) | Subject-specific interaction predictions L. iners prevents growth of other Lactobacillus spp. and L. jensenii aids the growth of Gardnerella sp. Finegoldia sp. have a highly important position in the vaginal microbiome and synergistic relationships with Sneathia and Anarococcus sp. L. iners was found to promote growth of Gardnerella as well as to promote growth of Atopobium, Prevotella, Parvimonas, Sneathia, and Mobiluncus |
| [114,165] | The longitudinal study included analysis of 16S amplicon sequencing and the Nugent score for vaginal samples | Mixed effects model Dynamic Bayesian network | L. iners and Streptococcus taxa are linked to menstrual cycle Found positive relationships between L. iners and Atopobium as well as Atopobium and Gardnerella |
| MOMS-PI dataset metatranscriptomic and metagenomic analysis of 122 vaginal samples | |||
| [160] | Integrated taxonomic and metabolomic data | Community-based metabolite potential (CMP) score | Association of specific metabolites and functional pathways to either healthy vaginal microbiomes or those with BV |
| [161] | Integrated metabolomic and taxonomic data collected from healthy women and women with BV, vulvovaginal candidiasis, and Chlamydia trachomatis infection | Co-abundance network of Spearman correlation coefficient | Lactobacillus spp. abundance was positively associated with lactate and 4-hydroxyphenylacetate, isoleucine, leucine, tryptophan, phenylalanine, and aspartate Lactobacillus was negatively correlated with formate, acetate, 2-hydroxyisovalerate, and alanine In contrast, other bacterial taxa were positively correlated with the metabolites that Lactobacillus was negatively correlated with; these include Gardnerella, Prevotella, Megasphaera, Atopobium, Dialister, and Clostridium. These taxa also showed a positive correlation with organic acids and amines |
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Unemo, M.; Seifert, H.S.; Hook, E.W.; Hawkes, S.; Ndowa, F.; Dillon, J.-A.R. Gonorrhoea. Nat. Rev. Dis. Prim. 2019, 5, 79. [Google Scholar] [CrossRef]
- Rowley, J.; Vander Hoorn, S.; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548–562. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2017; Available online: https://www.cdc.gov/std/stats17/2017-STD-Surveillance-Report_CDC-clearance-9.10.18.pdf (accessed on 16 March 2022).
- Apicella, M.A.; Ketterer, M.; Lee, F.K.N.; Zhou, D.; Rice, P.A.; Blake, M.S. The Pathogenesis of Gonococcal Urethritis in Men: Confocal and Immunoelectron Microscopic Analysis of Urethral Exudates from Men Infected with Neisseria gonorrhoeae. J. Infect. Dis. 1996, 173, 636–646. [Google Scholar] [CrossRef]
- Farzadegan, H.; Roth, I.L. Scanning electron microscopy and freeze-etching of gonorrhoeal urethral exudate. Sex. Transm. Infect. 1975, 51, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Katherine, H.; Ram, S.; Darville, T. Chapter 219 Neisseria gonorrhoeae (Gonococcus). In Nelson Textbook of Pediatrics; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Hook, E.W.; Holmes, K.K. Gonococcal Infections. Ann. Intern. Med. 1985, 102, 229–243. [Google Scholar] [CrossRef]
- Edwards, J.L.; Apicella, M.A. The Molecular Mechanisms Used by Neisseria gonorrhoeae To Initiate Infection Differ between Men and Women. Clin. Microbiol. Rev. 2004, 17, 965–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detels, R.; Green, A.M.; Klausner, J.D.; Katzenstein, D.; Gaydos, C.; Handsfield, H.H.; Pequegnat, W.; Mayer, K.; Hartwell, T.D.; Quinn, T.C. The Incidence and Correlates of Symptomatic and Asymptomatic Chlamydia trachomatis and Neisseria gonorrhoeae Infections in Selected Populations in Five Countries. Sex Transm Dis 2011, 38, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.L.; Butler, E.K. The Pathobiology of Neisseria gonorrhoeae Lower Female Genital Tract Infection. Front. Microbiol. 2011, 2, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesenfeld, H.C.; Hillier, S.L.; Meyn, L.A.; Amortegui, A.J.; Sweet, R.L. Subclinical Pelvic Inflammatory Disease and Infertility. Obstet. Gynecol. 2012, 120, 37–43. [Google Scholar] [CrossRef]
- Wu, H.; Soler-García, A.A.; Jerse, A.E. A Strain-Specific Catalase Mutation and Mutation of the Metal-Binding Transporter Gene mntC Attenuate Neisseria gonorrhoeae In Vivo but Not by Increasing Susceptibility to Oxidative Killing by Phagocytes. Infect. Immun. 2009, 77, 1091–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Buono, S.; Katz, K.A.; Pandori, M.W. Clinical Neisseria gonorrhoeae Isolates in the United States with Resistance to Azithromycin Possess Mutations in All 23S rRNA Alleles and the mtrR Coding Region. Microb. Drug Resist. 2011, 17, 425–427. [Google Scholar] [CrossRef]
- Nguyen, D.; Gose, S.; Castro, L.; Chung, K.; Bernstein, K.; Samuel, M.; Bauer, H.; Pandori, M. Neisseria gonorrhoeae Strain with Reduced Susceptibilities to Extended-Spectrum Cephalosporins. Emerg. Infect. Dis. 2014, 20, 1211. [Google Scholar] [CrossRef] [Green Version]
- Su, X.-H.; Wang, B.-X.; Le, W.-J.; Liu, Y.-R.; Wan, C.; Li, S.; Alm, R.A.; Mueller, J.P.; Rice, P.A. Multidrug-Resistant Neisseria gonorrhoeae Isolates from Nanjing, China, Are Sensitive to Killing by a Novel DNA Gyrase Inhibitor, ETX0914 (AZD0914). Antimicrob. Agents Chemother. 2016, 60, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.; Nemes-Nikodem, E.; Jeney, C.; Szabo, D.; Marschalko, M.; Karpati, S.; Ostorhazi, E. Emerging azithromycin-resistance among the Neisseria gonorrhoeae strains isolated in Hungary. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-W.; Li, L.-H.; Su, C.-Y.; Li, S.-Y.; Yen, M.-Y. Changes in the six most common sequence types of Neisseria gonorrhoeae, including ST4378, identified by surveillance of antimicrobial resistance in northern Taiwan from 2006 to 2013. J. Microbiol. Immunol. Infect. 2016, 49, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Unemo, M.; del Rio, C.; Shafer, W.M. Antimicrobial Resistance Expressed by Neisseria gonorrhoeae: A Major Global Public Health Problem in the 21st Century. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.A.; Robinette, A.; Montgomery, M.; Almonte, A.; Cu-Uvin, S.; Lonks, J.R.; Chapin, K.C.; Kojic, E.M.; Hardy, E.J. Extragenital Infections Caused by Chlamydia trachomatis and Neisseria gonorrhoeae: A Review of the Literature. Infect. Dis. Obstet. Gynecol. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Anahtar, M.N.; Byrne, E.H.; Doherty, K.E.; Bowman, B.A.; Yamamoto, H.S.; Soumillon, M.; Padavattan, N.; Ismail, N.; Moodley, A.; Sabatini, M.E.; et al. Cervicovaginal Bacteria Are a Major Modulator of Host Inflammatory Responses in the Female Genital Tract. Immunity 2015, 42, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, C.; Yang, W.; Wei, X.; Wu, K.; Huang, D. The Unique Microbiome and Innate Immunity During Pregnancy. Front. Immunol. 2019, 10, 2886. [Google Scholar] [CrossRef] [PubMed]
- Wira, C.R.; Grant-Tschudy, K.S.; Crane-Godreau, M.A. Epithelial Cells in the Female Reproductive Tract: A Central Role as Sentinels of Immune Protection. Am. J. Reprod. Immunol. 2005, 53, 65–76. [Google Scholar] [CrossRef]
- Wira, C.R.; Fahey, J.V.; Sentman, C.L.; Pioli, P.A.; Shen, L. Innate and adaptive immunity in female genital tract: Cellular responses and interactions. Immunol. Rev. 2005, 206, 306–335. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M. Hrsg. Antimicrobial Peptides and Human Disease: With 4 Tables; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Hickey, D.; Patel, M.; Fahey, J.; Wira, C. Innate and adaptive immunity at mucosal surfaces of the female reproductive tract: Stratification and integration of immune protection against the transmission of sexually transmitted infections. J. Reprod. Immunol. 2011, 88, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotman, E.; Seifert, H.S. The Genetics of Neisseria Species. Annu. Rev. Genet. 2014, 48, 405–431. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.P.; Meyer, T.F. Genome plasticity in Neisseria gonorrhoeae. FEMS Microbiol. Lett. 1996, 145, 173–179. [Google Scholar] [CrossRef]
- Masters, T.L.; Wachter, J.; Hill, S.A. Loop structures in the 5′ untranslated region and antisense RNA mediate pilE gene expression in Neisseria gonorrhoeae. Microbiology 2016, 162, 2005–2016. [Google Scholar] [CrossRef]
- Quillin, S.J.; Seifert, H.S. Neisseria gonorrhoeae host adaptation and pathogenesis. Nat. Rev. Genet. 2018, 16, 226–240. [Google Scholar] [CrossRef]
- Hill, S.A.; Samuels, D.S.; Carlson, J.H.; Wilson, J.; Hogan, D.; Lubke, L.; Belland, R.J. Integration host factor is a transcriptional cofactor of pilE in Neisseria gonorrhoeae. Mol. Microbiol. 1997, 23, 649–656. [Google Scholar] [CrossRef]
- McClure, R.; Sunkavalli, A.; Balzano, P.M.; Massari, P.; Cho, C.; Nauseef, W.M.; Apicella, M.A.; Genco, C.A. Global Network Analysis of Neisseria gonorrhoeae Identifies Coordination between Pathways, Processes, and Regulators Expressed during Human Infection. mSystems 2020, 5, e00729-19. [Google Scholar] [CrossRef] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.D.; Skaar, E.P. Transition Metals and Virulence in Bacteria. Annu. Rev. Genet. 2016, 50, 67–91. [Google Scholar] [CrossRef] [Green Version]
- Hennigar, S.R.; McClung, J.P. Nutritional Immunity. Am. J. Lifestyle Med. 2016, 10, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Liu, C.; Mitchell, C.M.; Fiedler, T.L.; Thomas, K.K.; Agnew, K.J.; Marrazzo, J.; Fredricks, D.N. Temporal Variability of Human Vaginal Bacteria and Relationship with Bacterial Vaginosis. PLoS ONE 2010, 5, e10197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.A.; Brabin, L.; Diallo, S.; Gies, S.; Nelson, A.; Stewart, C.; Swinkels, D.W.; Geurts-Moespot, A.J.; Kazienga, A.; Ouedraogo, S.; et al. Mucosal lactoferrin response to genital tract infections is associated with iron and nutritional biomarkers in young Burkinabé women. Eur. J. Clin. Nutr. 2019, 73, 1464–1472. [Google Scholar] [CrossRef]
- Cornelissen, C.N. Subversion of nutritional immunity by the pathogenic Neisseriae. Pathog. Dis. 2017, 76, ftx112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, E.D. Iron availability and infection. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Cornelis, P.; Wei, Q.; Andrews, S.C.; Vinckx, T. Iron homeostasis and management of oxidative stress response in bacteria. Metallomics 2011, 3, 540–549. [Google Scholar] [CrossRef]
- Yu, C.; McClure, R.; Nudel, K.; Daou, N.; Genco, C.A. Characterization of the Neisseria gonorrhoeae Iron and Fur Regulatory Network. J. Bacteriol. 2016, 198, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Hollander, A.; Mercante, A.D.; Shafer, W.M.; Cornelissen, C.N. The Iron-Repressed, AraC-Like Regulator MpeR Activates Expression of fetA in Neisseria gonorrhoeae. Infect. Immun. 2011, 79, 4764–4776. [Google Scholar] [CrossRef] [Green Version]
- Nudel, K.; McClure, R.; Moreau, M.; Briars, E.; Abrams, A.J.; Tjaden, B.; Su, X.-H.; Trees, D.; Rice, P.A.; Massari, P.; et al. Transcriptome Analysis of Neisseria gonorrhoeae during Natural Infection Reveals Differential Expression of Antibiotic Resistance Determinants between Men and Women. mSphere 2018, 3, e00312-18. [Google Scholar] [CrossRef] [Green Version]
- Folster, J.P.; Shafer, W.M. Regulation of mtrF Expression in Neisseria gonorrhoeae and Its Role in High-Level Antimicrobial Resistance. J. Bacteriol. 2005, 187, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Fillat, M.F. The FUR (ferric uptake regulator) superfamily: Diversity and versatility of key transcriptional regulators. Arch. Biochem. Biophys. 2014, 546, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Bagg, A.; Neilands, J.B. Ferric uptake regulation protein acts as a repressor, employing iron(II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 1987, 26, 5471–5477. [Google Scholar] [CrossRef]
- Sheikh, A.; Taylor, G.L. Crystal structure of the Vibrio cholerae ferric uptake regulator (Fur) reveals insights into metal co-ordination. Mol. Microbiol. 2009, 72, 1208–1220. [Google Scholar] [CrossRef]
- Yu, C.; Genco, C.A. Fur-Mediated Global Regulatory Circuits in Pathogenic Neisseria Species. J. Bacteriol. 2012, 194, 6372–6381. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.M.; Gilbreath, J.J.; Pich, O.Q.; McKelvey, A.M.; Maynard, E.L.; Li, Z.-Z.; Merrell, D.S. Identification and Characterization of Novel Helicobacter pylori apo-Fur-Regulated Target Genes. J. Bacteriol. 2013, 195, 5526–5539. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.M.; Whitmire, J.M.; Merrell, D.S. This Is Not Your Mother’s Repressor: The Complex Role of Fur in Pathogenesis. Infect. Immun. 2009, 77, 2590–2601. [Google Scholar] [CrossRef] [Green Version]
- Jackson, L.A.; Ducey, T.F.; Day, M.W.; Zaitshik, J.B.; Orvis, J.; Dyer, D.W. Transcriptional and Functional Analysis of the Neisseria gonorrhoeae Fur Regulon. J. Bacteriol. 2010, 192, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, C.N.; Hollander, A. TonB-Dependent Transporters Expressed by Neisseria gonorrhoeae. Front. Microbiol. 2011, 2, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, C.N.; Kelley, M.; Hobbs, M.M.; Anderson, J.E.; Cannon, J.G.; Cohen, M.S.; Sparling, P.F. The transferrin receptor expressed by gonococcal strain FA1090 is required for the experimental infection of human male volunteers. Mol. Microbiol. 1998, 27, 611–616. [Google Scholar] [CrossRef]
- Cornelissen, C.N.; Biswas, G.D.; Tsai, J.; Paruchuri, D.K.; Thompson, S.A.; Sparling, P.F. Gonococcal transferrin-binding protein 1 is required for transferrin utilization and is homologous to TonB-dependent outer membrane receptors. J. Bacteriol. 1992, 174, 5788–5797. [Google Scholar] [CrossRef] [Green Version]
- Ducey, T.F.; Carson, M.B.; Orvis, J.; Stintzi, A.P.; Dyer, D.W. Identification of the Iron-Responsive Genes of Neisseria gonorrhoeae by Microarray Analysis in Defined Medium. J. Bacteriol. 2005, 187, 4865–4874. [Google Scholar] [CrossRef] [Green Version]
- Ducey, T.F.; Jackson, L.; Orvis, J.; Dyer, D.W. Transcript analysis of nrrF, a Fur repressed sRNA of Neisseria gonorrhoeae. Microb. Pathog. 2009, 46, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Mellin, J.R.; Goswami, S.; Grogan, S.; Tjaden, B.; Genco, C.A. A Novel Fur- and Iron-Regulated Small RNA, NrrF, Is Required for Indirect Fur-Mediated Regulation of the sdhA and sdhC Genes in Neisseria meningitidis. J. Bacteriol. 2007, 189, 3686–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Gallegos, M.T.; Schleif, R.; Bairoch, A.; Hofmann, K.; Ramos, J.L. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 1997, 61, 393–410. [Google Scholar]
- Mercante, A.D.; Jackson, L.; Johnson, P.J.T.; Stringer, V.A.; Dyer, D.W.; Shafer, W.M. MpeR Regulates the mtr Efflux Locus in Neisseria gonorrhoeae and Modulates Antimicrobial Resistance by an Iron-Responsive Mechanism. Antimicrob. Agents Chemother. 2012, 56, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Avalos, D.; Martínez-Pérez, N.; Ortiz-Moncada, M.A.; Juárez-González, A.; Baños-Vargas, A.A.; Santos, P.E.-D.L.; Pérez-Rueda, E.; Ibarra, J.A. An update of the unceasingly growing and diverse AraC/XylS family of transcriptional activators. FEMS Microbiol. Rev. 2021, 45, fuab020. [Google Scholar] [CrossRef]
- Quillin, S.J.; Hockenberry, A.J.; Jewett, M.C.; Seifert, H.S. Neisseria gonorrhoeae Exposed to Sublethal Levels of Hydrogen Peroxide Mounts a Complex Transcriptional Response. mSystems 2018, 3, e00156-18. [Google Scholar] [CrossRef] [Green Version]
- Stohl, E.A.; Criss, A.K.; Seifert, H.S. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 2005, 58, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Zughaier, S.M.; Kandler, J.L.; Shafer, W.M. Neisseria gonorrhoeae Modulates Iron-Limiting Innate Immune Defenses in Macrophages. PLoS ONE 2014, 9, e87688. [Google Scholar] [CrossRef]
- Becker, K.W.; Skaar, E.P. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol. Rev. 2014, 38, 1235–1249. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Seib, K.; Srikhanta, Y.; Kidd, S.; Edwards, J.L.; Maguire, T.L.; Grimmond, S.; Apicella, M.A.; McEwan, A.G.; Jennings, M.P. PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol. Microbiol. 2006, 60, 401–416. [Google Scholar] [CrossRef]
- Jean, S.; Juneau, R.A.; Criss, A.K.; Cornelissen, C.N. Neisseria gonorrhoeae Evades Calprotectin-Mediated Nutritional Immunity and Survives Neutrophil Extracellular Traps by Production of TdfH. Infect. Immun. 2016, 84, 2982–2994. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.P.; Potter, A.J.; Apicella, M.A.; Jennings, M.P.; McEwan, A.G. NmlR of Neisseria gonorrhoeae: A novel redox responsive transcription factor from the MerR family: Neisseria MerR-like transcription factor. Mol. Microbiol. 2005, 57, 1676–1689. [Google Scholar] [CrossRef]
- Lim, K.H.L.; Jones, C.E.; Hoven, R.N.V.; Edwards, J.L.; Falsetta, M.L.; Apicella, M.A.; Jennings, M.P.; McEwan, A.G. Metal Binding Specificity of the MntABC Permease of Neisseria gonorrhoeae and Its Influence on Bacterial Growth and Interaction with Cervical Epithelial Cells. Infect. Immun. 2008, 76, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Seib, K.L.; Srikhanta, Y.N.; Edwards, J.; Kidd, S.P.; Maguire, T.L.; Hamilton, A.; Pan, K.-T.; Hsiao, H.-H.; Yao, C.-W.; et al. Manganese regulation of virulence factors and oxidative stress resistance in Neisseria gonorrhoeae. J. Proteom. 2010, 73, 899–916. [Google Scholar] [CrossRef] [Green Version]
- Helmann, J.D.; Foster, A.W.; Osman, D.; Robinson, N.J. Specificity of Metal Sensing: Iron and Manganese Homeostasis in Bacillus subtilis. J. Biol. Chem. 2014, 289, 28112–28120. [Google Scholar] [CrossRef] [Green Version]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen–host interface. Nat. Rev. Genet. 2012, 10, 525–537. [Google Scholar] [CrossRef]
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.; Wu, H.-J.; Srikhanta, Y.; Edwards, J.L.; Falsetta, M.L.; Hamilton, A.J.; Maguire, T.L.; Grimmond, S.; Apicella, M.A.; McEwan, A.G.; et al. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol. Microbiol. 2007, 63, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-J.; McEwan, A.G.; Apicella, M.A.; Jennings, M.P. OxyR Acts as a Repressor of Catalase Expression in Neisseria gonorrhoeae. Infect. Immun. 2003, 71, 550–556. [Google Scholar] [CrossRef] [Green Version]
- Christman, M.F.; Storz, G.; Ames, B.N. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 3484–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schook, P.O.P.; Stohl, E.A.; Criss, A.K.; Seifert, H.S. The DNA-binding activity of the Neisseria gonorrhoeae LexA orthologue NG1427 is modulated by oxidation. Mol. Microbiol. 2010, 79, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Newkirk, G.R. Pelvic inflammatory disease: A contemporary approach. Am. Fam. Physician 1996, 53, 1127–1135. [Google Scholar]
- Clark, V.L.; Knapp, J.S.; Thompson, S.; Klimpel, K.W. Presence of antibodies to the major anaerobically induced gonococcal outer membrane protein in sera from patients with gonococcal infections. Microb. Pathog. 1988, 5, 381–390. [Google Scholar] [CrossRef]
- Barth, K.R.; Isabella, V.M.; Clark, V.L. Biochemical and genomic analysis of the denitrification pathway within the genus Neisseria. Microbiology 2009, 155, 4093–4103. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.M.; Clark, V.L. Deep sequencing-based analysis of the anaerobic stimulon in Neisseria gonorrhoeae. BMC Genom. 2011, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Lissenden, S.; Mohan, S.; Overton, T.; Regan, T.; Crooke, H.; Cardinale, J.A.; Householder, T.C.; Adams, P.; O’Conner, C.D.; Clark, V.L.; et al. Identification of transcription activators that regulate gonococcal adaptation from aerobic to anaerobic or oxygen-limited growth. Mol. Microbiol. 2000, 37, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Overton, T.; Whitehead, R.; Li, Y.; Snyder, L.; Saunders, N.; Smith, H.; Cole, J.A. Coordinated Regulation of the Neisseria gonorrhoeae-truncated Denitrification Pathway by the Nitric Oxide-sensitive Repressor, NsrR, and Nitrite-insensitive NarQ-NarP. J. Biol. Chem. 2006, 281, 33115–33126. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, R.N.; Overton, T.W.; Snyder, L.A.S.; McGowan, S.J.; Smith, H.; Cole, J.A.; Saunders, N.J. The small FNR regulon of Neisseria gonorrhoeae: Comparison with the larger Escherichia coli FNR regulon and interaction with the NarQ-NarP regulon. BMC Genom. 2007, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.; Wright, L.F.; Barth, K.; Spence, J.M.; Grogan, S.; Genco, C.A.; Clark, V.L. cis- and trans-acting elements involved in regulation of norB (norZ), the gene encoding nitric oxide reductase in Neisseria gonorrhoeae. Microbiology 2008, 154, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.M.; Jr, J.D.L.; Kennedy, E.M.; Clark, V.L. Functional analysis of NsrR, a nitric oxide-sensing Rrf2 repressor in Neisseria gonorrhoeae. Mol. Microbiol. 2008, 71, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Woude, M.W.; Bäumler, A.J. Phase and Antigenic Variation in Bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, C.D.; Palmer, M.E. Evolution of simple sequence repeat-mediated phase variation in bacterial genomes. Ann. N. Y. Acad. Sci. 2012, 1267, 39–44. [Google Scholar] [CrossRef]
- Jordan, P.W.; Snyder, L.A.; Saunders, N.J. Strain-specific differences in Neisseria gonorrhoeae associated with the phase variable gene repertoire. BMC Microbiol. 2005, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Zelewska, M.A.; Pulijala, M.; Spencer-Smith, R.; Mahmood, H.-T.A.; Norman, B.; Churchward, C.; Calder, A.; Snyder, L.A.S. Phase variable DNA repeats in Neisseria gonorrhoeae influence transcription, translation, and protein sequence variation. Microb. Genom. 2016, 2, e000078. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Atack, J.M.; Jennings, M.P.; Seib, K.L. The Capricious Nature of Bacterial Pathogens: Phasevarions and Vaccine Development. Front. Immunol. 2016, 7, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, S.; Stone, B.; Beucher, M.; Fu, J.; Sparling, P.F. Phase variation of the gonococcal siderophore receptor FetA. Mol. Microbiol. 2002, 36, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.L.; Srikhanta, Y.N.; Atack, J.M.; Jennings, M.P. Epigenetic Regulation of Virulence and Immunoevasion by Phase-Variable Restriction-Modification Systems in Bacterial Pathogens. Annu. Rev. Microbiol. 2020, 74, 655–671. [Google Scholar] [CrossRef]
- Srikhanta, Y.; Dowideit, S.J.; Edwards, J.L.; Falsetta, M.L.; Wu, H.-J.; Harrison, O.; Fox, K.L.; Seib, K.; Maguire, T.L.; Wang, A.H.-J.; et al. Phasevarions Mediate Random Switching of Gene Expression in Pathogenic Neisseria. PLOS Pathog. 2009, 5, e1000400. [Google Scholar] [CrossRef] [Green Version]
- Srikhanta, Y.N.; Fox, K.L.; Jennings, M.P. The phasevarion: Phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Genet. 2010, 8, 196–206. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-Scale Mapping and Validation of Escherichia coli Transcriptional Regulation from a Compendium of Expression Profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Saint-André, V. Computational biology approaches for mapping transcriptional regulatory networks. Comput. Struct. Biotechnol. J. 2021, 19, 4884–4895. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; Van Der Graaf, A.; Franke, L.; De Magalhães, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Briefings Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- McDermott, J.E.; Taylor, R.C.; Yoon, H.; Heffron, F. Bottlenecks and Hubs in Inferred Networks Are Important for Virulence in Salmonella typhimurium. J. Comput. Biol. 2009, 16, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Ye, X.; Li, C.; Wang, Y. K-Module Algorithm: An Additional Step to Improve the Clustering Results of WGCNA Co-Expression Networks. Genes 2021, 12, 87. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Sanguinetti, G. Gene Regulatory Network Inference: An Introductory Survey. In Gene Regulatory Networks; Sanguinetti, G., Huynh-Thu, V.A., Eds.; Springer: New York, NY, USA, 2019; pp. 1–23. [Google Scholar]
- Gillis, J.; Pavlidis, P. “Guilt by Association” Is the Exception Rather Than the Rule in Gene Networks. PLoS Comput. Biol. 2012, 8, e1002444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, F.M.T.; Bernstein, K.T.; Aral, S.O. Vaginal Microbiome and Its Relationship to Behavior, Sexual Health, and Sexually Transmitted Diseases. Obstet. Gynecol. 2017, 129, 643–654. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4680–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. npj Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finotello, F.; Mastrorilli, E.; Di Camillo, B. Measuring the diversity of the human microbiota with targeted next-generation sequencing. Briefings Bioinform. 2016, 19, 679–692. [Google Scholar] [CrossRef]
- Sycuro, L.K.; Fredricks, D.N. Microbiota of the Genitourinary Tract. In The Human Microbiota; Fredricks, D.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 167–210. [Google Scholar]
- Chen, C.; Song, X.; Chunwei, Z.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.M.; Haick, A.; Nkwopara, E.; Garcia, R.; Rendi, M.; Agnew, K.; Fredricks, D.; Eschenbach, D. Colonization of the upper genital tract by vaginal bacterial species in nonpregnant women. Am. J. Obstet. Gynecol. 2014, 212, 611.e1–611.e9. [Google Scholar] [CrossRef] [Green Version]
- Aldunate, M.; Srbinovski, D.; Hearps, A.C.; Latham, C.F.; Ramsland, P.A.; Gugasyan, R.; Cone, R.A.; Tachedjian, G. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front. Physiol. 2015, 6, 164. [Google Scholar] [CrossRef] [PubMed]
- Plesniarski, A.; Siddik, A.B.; Su, R.-C. The Microbiome as a Key Regulator of Female Genital Tract Barrier Function. Front. Cell. Infect. Microbiol. 2021, 11, 790627. [Google Scholar] [CrossRef]
- Zhou, X.; Bent, S.J.; Schneider, M.G.; Davis, C.C.; Islam, M.R.; Forney, L.J. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 2004, 150, 2565–2573. [Google Scholar] [CrossRef] [Green Version]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schütte, U.M.E.; Zhong, X.; Koenig, S.S.K.; Fu, L.; Ma, Z.; Zhou, X.; et al. Temporal Dynamics of the Human Vaginal Microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peric, A.; Weiss, J.; Vulliemoz, N.; Baud, D.; Stojanov, M. Bacterial Colonization of the Female Upper Genital Tract. Int. J. Mol. Sci. 2019, 20, 3405. [Google Scholar] [CrossRef] [Green Version]
- Miles, S.M.; Hardy, B.L.; Merrell, D. Investigation of the microbiota of the reproductive tract in women undergoing a total hysterectomy and bilateral salpingo-oopherectomy. Fertil. Steril. 2017, 107, 813–820.e1. [Google Scholar] [CrossRef] [Green Version]
- Franasiak, J.M.; Werner, M.D.; Juneau, C.R.; Tao, X.; Landis, J.; Zhan, Y.; Treff, N.R.; Scott, R.T. Endometrial microbiome at the time of embryo transfer: Next-generation sequencing of the 16S ribosomal subunit. J. Assist. Reprod. Genet. 2016, 33, 129–136. [Google Scholar] [CrossRef]
- Moreno, I.; Codoñer, F.M.; Vilella, F.; Valbuena, D.; Martinez-Blanch, J.F.; Jimenez-Almazán, J.; Alonso, R.; Alamá, P.; Remohí, J.; Pellicer, A.; et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am. J. Obstet. Gynecol. 2016, 215, 684–703. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, E.S.; Willner, D.; Buttini, M.; Hafner, L.M.; Theodoropoulos, C.; Huygens, F. The fallopian tube microbiome: Implications for reproductive health. Oncotarget 2018, 9, 21541–21551. [Google Scholar] [CrossRef]
- Zhou, B.; Sun, C.; Huang, J.; Xia, M.; Guo, E.; Li, N.; Lu, H.; Shan, W.; Wu, Y.; Li, Y.; et al. The biodiversity Composition of Microbiome in Ovarian Carcinoma Patients. Sci. Rep. 2019, 9, 1691. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, R.; He, W.; Zhong, X.; Liu, W.; Zhu, H.; Zhang, X.; Luo, Q. Modulation effect of vaginal mucosal microflora and susceptibility to Neisseria gonorrhoeae infections: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2019, 300, 261–267. [Google Scholar] [CrossRef]
- Kovachev, S. Defence factors of vaginal lactobacilli. Crit. Rev. Microbiol. 2017, 44, 31–39. [Google Scholar] [CrossRef]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Foschi, C.; Salvo, M.; Cevenini, R.; Parolin, C.; Vitali, B.; Marangoni, A. Vaginal Lactobacilli Reduce Neisseria gonorrhoeae Viability through Multiple Strategies: An in Vitro Study. Front. Cell. Infect. Microbiol. 2017, 7, 502. [Google Scholar] [CrossRef]
- Spurbeck, R.R.; Arvidson, C.G. Inhibition of Neisseria gonorrhoeae Epithelial Cell Interactions by Vaginal Lactobacillus Species. Infect. Immun. 2008, 76, 3124–3130. [Google Scholar] [CrossRef] [Green Version]
- Spurbeck, R.R.; Arvidson, C.G. Lactobacillus jensenii Surface-Associated Proteins Inhibit Neisseria gonorrhoeae Adherence to Epithelial Cells. Infect. Immun. 2010, 78, 3103–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainulainen, V.; Loimaranta, V.; Pekkala, A.; Edelman, S.; Antikainen, J.; Kylvaja, R.; Laaksonen, M.; Laakkonen, L.; Finne, J.; Korhonen, T.K. Glutamine Synthetase and Glucose-6-Phosphate Isomerase Are Adhesive Moonlighting Proteins of Lactobacillus crispatus Released by Epithelial Cathelicidin LL-37. J. Bacteriol. 2012, 194, 2509–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Płaczkiewicz, J.; Chmiel, P.; Malinowska, E.; Bącal, P.; Kwiatek, A. Lactobacillus crispatus and its enolase and glutamine synthetase influence interactions between Neisseria gonorrhoeae and human epithelial cells. J. Microbiol. 2020, 58, 405–414. [Google Scholar] [CrossRef]
- Cook, R.L.; Reid, G.; Pond, D.G.; Schmitt, C.A.; Sobel, J.D. Clue Cells in Bacterial Vaginosis: Immunofluorescent Identification of the Adherent Gram-Negative Bacteria as Gardnerella vaginalis. J. Infect. Dis. 1989, 160, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Ser, H.-L.; Kuai, Y.-H.; Tan, L.; Arasoo, V.; Letchumanan, V.; Wang, L.; Pusparajah, P.; Goh, B.-H.; Ab Mutalib, N.-S.; et al. Finding a Balance in the Vaginal Microbiome: How Do We Treat and Prevent the Occurrence of Bacterial Vaginosis? Antibiotics 2021, 10, 719. [Google Scholar] [CrossRef]
- Redelinghuys, M.J.; Geldenhuys, J.; Jung, H.; Kock, M.M. Bacterial Vaginosis: Current Diagnostic Avenues and Future Opportunities. Front. Cell. Infect. Microbiol. 2020, 10, 354. [Google Scholar] [CrossRef]
- Pettit, R.K.; McAllister, S.C.; Hamer, T.A. Response of gonococcal clinical isolates to acidic conditions. J. Med. Microbiol. 1999, 48, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Pettit, R.; Whelan, T.; Woo, K. Acid stress upregulated outer membrane proteins in clinical isolates of Neisseria gonorrhoeae, but not most commensal Neisseria. Can. J. Microbiol. 2001, 47, 871–876. [Google Scholar] [CrossRef]
- Rice, P.A.; Shafer, W.M.; Ram, S.; Jerse, A.E. Neisseria gonorrhoeae: Drug Resistance, Mouse Models, and Vaccine Development. Annu. Rev. Microbiol. 2017, 71, 665–686. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Tang, M.M.; Wu, X.; Phillips, N.; Galkowski, D.; Jarvis, G.; Fan, H. Arginine- and Polyamine-Induced Lactic Acid Resistance in Neisseria gonorrhoeae. PLoS ONE 2016, 11, e0147637. [Google Scholar] [CrossRef]
- Mehta, S.D.; Zhao, D.; Green, S.J.; Agingu, W.; Otieno, F.; Bhaumik, R.; Bhaumik, D.; Bailey, R.C. The Microbiome Composition of a Man’s Penis Predicts Incident Bacterial Vaginosis in His Female Sex Partner with High Accuracy. Front. Cell. Infect. Microbiol. 2020, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Borovkova, N.; Korrovits, P.; Ausmees, K.; Türk, S.; Jõers, K.; Punab, M.; Mändar, R. Influence of sexual intercourse on genital tract microbiota in infertile couples. Anaerobe 2011, 17, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Mändar, R. Microbiota of male genital tract: Impact on the health of man and his partner. Pharmacol. Res. 2013, 69, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Aragón, I.M.; Herrera-Imbroda, B.; Queipo-Ortuño, M.I.; Castillo, E.; Del Moral, J.S.-G.; Gómez-Millán, J.; Yucel, G.; Lara, M.F. The Urinary Tract Microbiome in Health and Disease. Eur. Urol. Focus 2018, 4, 128–138. [Google Scholar] [CrossRef]
- Kim, M.S.; Jung, S.I. The Urinary Tract Microbiome in Male Genitourinary Diseases: Focusing on Benign Prostate Hyperplasia and Lower Urinary Tract Symptoms. Int. Neurourol. J. 2021, 25, 3–11. [Google Scholar] [CrossRef]
- Nelson, D.E.; Van Der Pol, B.; Dong, Q.; Revanna, K.V.; Fan, B.; Easwaran, S.; Sodergren, E.; Weinstock, G.M.; Diao, L.; Fortenberry, J.D. Characteristic Male Urine Microbiomes Associate with Asymptomatic Sexually Transmitted Infection. PLoS ONE 2010, 5, e14116. [Google Scholar] [CrossRef]
- Nelson, D.E.; Dong, Q.; Van Der Pol, B.; Toh, E.; Fan, B.; Katz, B.P.; Mi, D.; Rong, R.; Weinstock, G.M.; Sodergren, E.; et al. Bacterial Communities of the Coronal Sulcus and Distal Urethra of Adolescent Males. PLoS ONE 2012, 7, e36298. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Nelson, D.E.; Toh, E.; Diao, L.; Gao, X.; Fortenberry, J.D.; Van Der Pol, B. The Microbial Communities in Male First Catch Urine Are Highly Similar to Those in Paired Urethral Swab Specimens. PLoS ONE 2011, 6, e19709. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.-J.; Huang, S.-T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K.; et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Frølund, M.; Wikström, A.; Lidbrink, P.; Abu Al-Soud, W.; Larsen, N.; Harder, C.B.; Sørensen, S.J.; Jensen, J.S.; Ahrens, P. The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PLoS ONE 2018, 13, e0201380. [Google Scholar] [CrossRef] [Green Version]
- Rakhmatulina, M.R.; Boldyreva, M.N.; Lipova, E.V.; Chekmarev, A.S.; Galkina, I.S. Evaluation of the composition of the microbiota of the urethra in men with sexually transmitted infections. Urologiia 2019, 6, 31–37. [Google Scholar] [CrossRef]
- Saraf, V.S.; Sheikh, S.A.; Ahmad, A.; Gillevet, P.M.; Bokhari, H.; Javed, S. Vaginal microbiome: Normalcy vs dysbiosis. 2021, 203, 3793–3802. Arch. Microbiol. 2021, 203, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Munckhof, E.H.A.V.D.; van Sitter, R.L.; Boers, K.E.; Lamont, R.F.; Witt, R.T.; le Cessie, S.; Knetsch, C.W.; van Doorn, L.-J.; Quint, W.G.V.; Molijn, A.; et al. Comparison of Amsel criteria, Nugent score, culture and two CE-IVD marked quantitative real-time PCRs with microbiota analysis for the diagnosis of bacterial vaginosis. Eur. J. Clin. Microbiol. 2019, 38, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Nugent, R.P.; Krohn, M.A.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Gd, B.; Ca, H. Utility of Next-generation sequencing in Managing Bacterial Vaginosis: Examples from Clinical Practice. J. Women’s Health Care 2016, 5, 1–6. [Google Scholar] [CrossRef]
- Deng, Z.-L.; Gottschick, C.; Bhuju, S.; Masur, C.; Abels, C.; Wagner-Döbler, I. Metatranscriptome Analysis of the Vaginal Microbiota Reveals Potential Mechanisms for Protection against Metronidazole in Bacterial Vaginosis. mSphere 2018, 3, e00262-18. [Google Scholar] [CrossRef] [Green Version]
- Twin, J.; Bradshaw, C.S.; Garland, S.M.; Fairley, C.K.; Fethers, K.; Tabrizi, S.N. The Potential of Metatranscriptomics for Identifying Screening Targets for Bacterial Vaginosis. PLoS ONE 2013, 8, e76892. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ji, B.; Zengler, K.; Nielsen, J. Modelling approaches for studying the microbiome. Nat. Microbiol. 2019, 4, 1253–1267. [Google Scholar] [CrossRef]
- Lachance, J.; Matteau, D.; Brodeur, J.; Lloyd, C.J.; Mih, N.; King, Z.A.; Knight, T.F.; Feist, A.M.; Monk, J.M.; Palsson, B.O.; et al. Genome-scale metabolic modeling reveals key features of a minimal gene set. Mol. Syst. Biol. 2021, 17, e10099. [Google Scholar] [CrossRef]
- Buchweitz, L.F.; Yurkovich, J.T.; Blessing, C.; Kohler, V.; Schwarzkopf, F.; King, Z.A.; Yang, L.; Jóhannsson, F.; Sigurjónsson, Ó.E.; Rolfsson, Ó.; et al. Visualizing metabolic network dynamics through time-series metabolomic data. BMC Bioinform. 2020, 21, 130. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Fong, S.S. Computational Modeling of the Human Microbiome. Microorganisms 2020, 8, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, S.; Huang, B.; Parikh, H.I.; Edwards, D.J.; Brooks, J.P.; Kumar, N.G.; Sheth, N.U.; Koparde, V.; Smirnova, E.; Huzurbazar, S.; et al. Multi-omic Microbiome Profiles in the Female Reproductive Tract in Early Pregnancy. Infect. Microbes Dis. 2019, 1, 49–60. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.L.; Edwards, D.J.; Girerd, P.H.; Parikh, H.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.G.; Parikh, H.; Brooks, J.L.; Edwards, D.J.; Arodz, T.J.; Edupuganti, L.; Huang, B.; Girerd, P.H.; Bokhari, Y.A.; Bradley, S.P.; et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 2019, 25, 1001–1011. [Google Scholar] [CrossRef]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 2016, 1, e00013-15. [Google Scholar] [CrossRef] [Green Version]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [Green Version]
- Brotman, R.M.; Klebanoff, M.A.; Nansel, T.; Yu, K.F.; Andrews, W.W.; Zhang, J.; Schwebke, J.R. Bacterial Vaginosis Assessed by Gram Stain and Diminished Colonization Resistance to Incident Gonococcal, Chlamydial, and Trichomonal Genital Infection. J. Infect. Dis. 2010, 202, 1907–1915. [Google Scholar] [CrossRef]
- Brotman, R.M.; Bradford, L.L.; Conrad, M.; Gajer, P.; Ault, K.; Peralta, L.; Forney, L.J.; Carlton, J.M.; Abdo, Z.; Ravel, J. Association Between Trichomonas vaginalis and Vaginal Bacterial Community Composition Among Reproductive-Age Women. Sex Transm Dis 2012, 39, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.; Kelkar, Y.D.; Gu, Y.; Zhou, J.; Qiu, X.; Wu, H. High-dimensional linear state space models for dynamic microbial interaction networks. PLoS ONE 2017, 12, e0187822. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Martinez, J.; Ruiz-Perez, D.; Narasimhan, G.; Bar-Joseph, Z. Dynamic interaction network inference from longitudinal microbiome data. Microbiome 2019, 7, 54. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunkavalli, A.; McClure, R.; Genco, C. Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae. Microorganisms 2022, 10, 922. https://doi.org/10.3390/microorganisms10050922
Sunkavalli A, McClure R, Genco C. Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae. Microorganisms. 2022; 10(5):922. https://doi.org/10.3390/microorganisms10050922
Chicago/Turabian StyleSunkavalli, Ashwini, Ryan McClure, and Caroline Genco. 2022. "Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae" Microorganisms 10, no. 5: 922. https://doi.org/10.3390/microorganisms10050922
APA StyleSunkavalli, A., McClure, R., & Genco, C. (2022). Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae. Microorganisms, 10(5), 922. https://doi.org/10.3390/microorganisms10050922