
Microbial Interrelationships across Sites of Breastfeeding Mothers and Infants at 6 Weeks Postpartum




Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Study Design and Data Collection
2.3. Sample Collection
2.4. DNA Extraction
2.5. PCR Amplification and 16S rRNA Sequencing
2.6. Sequence Processing
2.7. Statistical Analyses
2.8. Microbial Source Prediction Analysis
2.9. Co-Occurrence Network Analysis
3. Results
3.1. Subject Characteristics
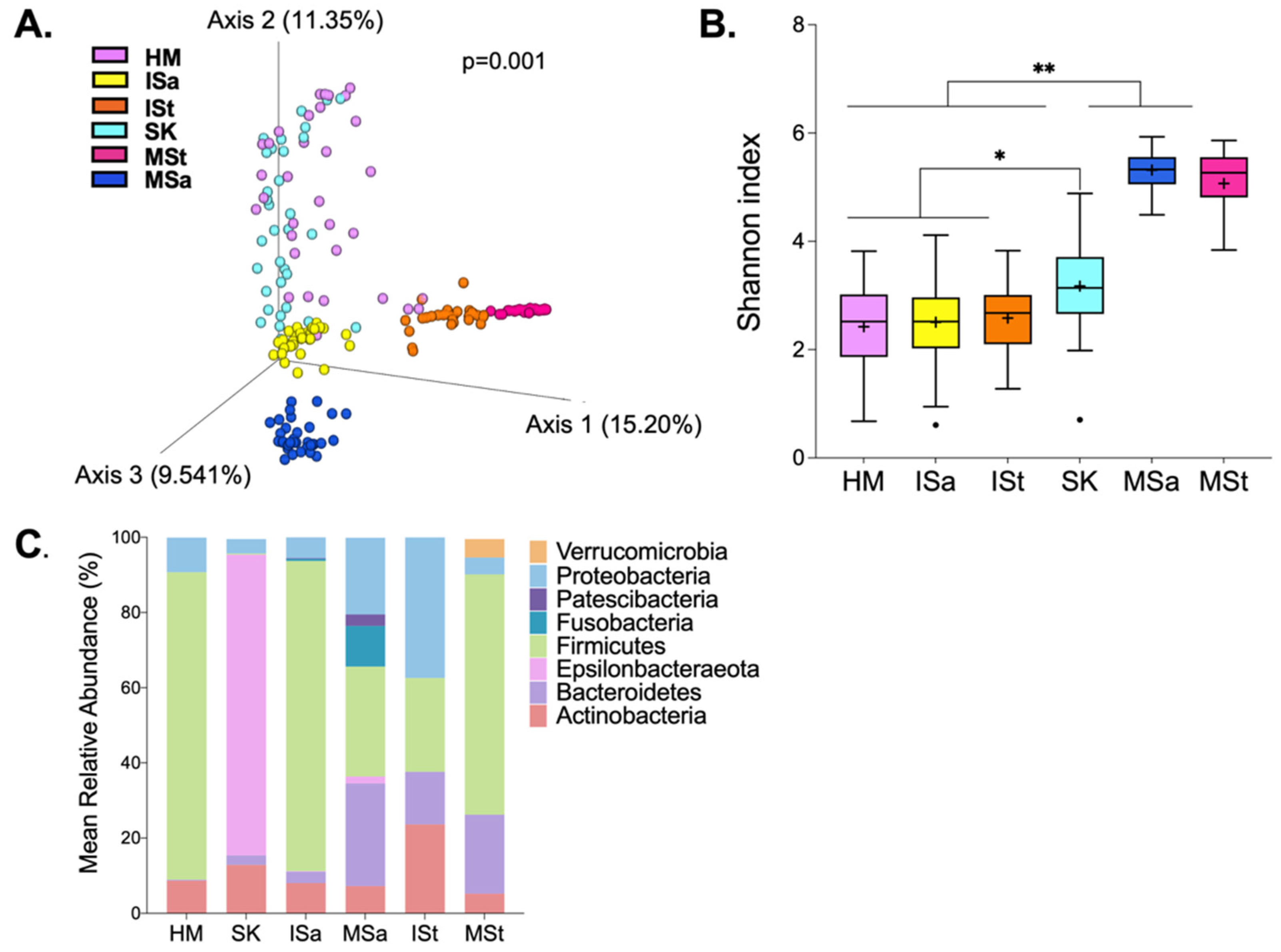
3.2. Bacterial Composition among Maternal and Infant Body Sites
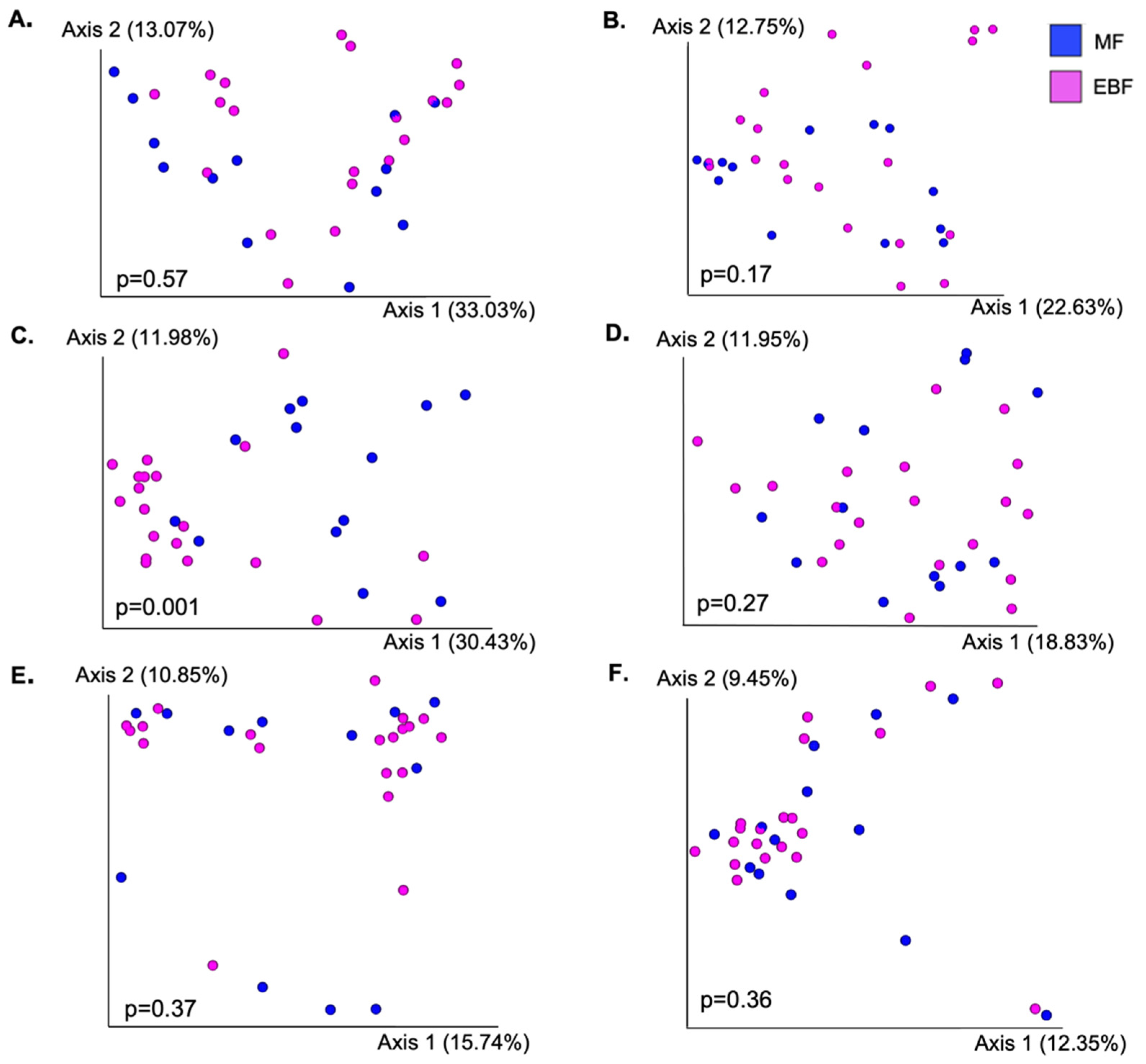
3.3. Bacterial Diversity between Feeding Groups
3.4. Taxonomic Composition between Feeding Groups
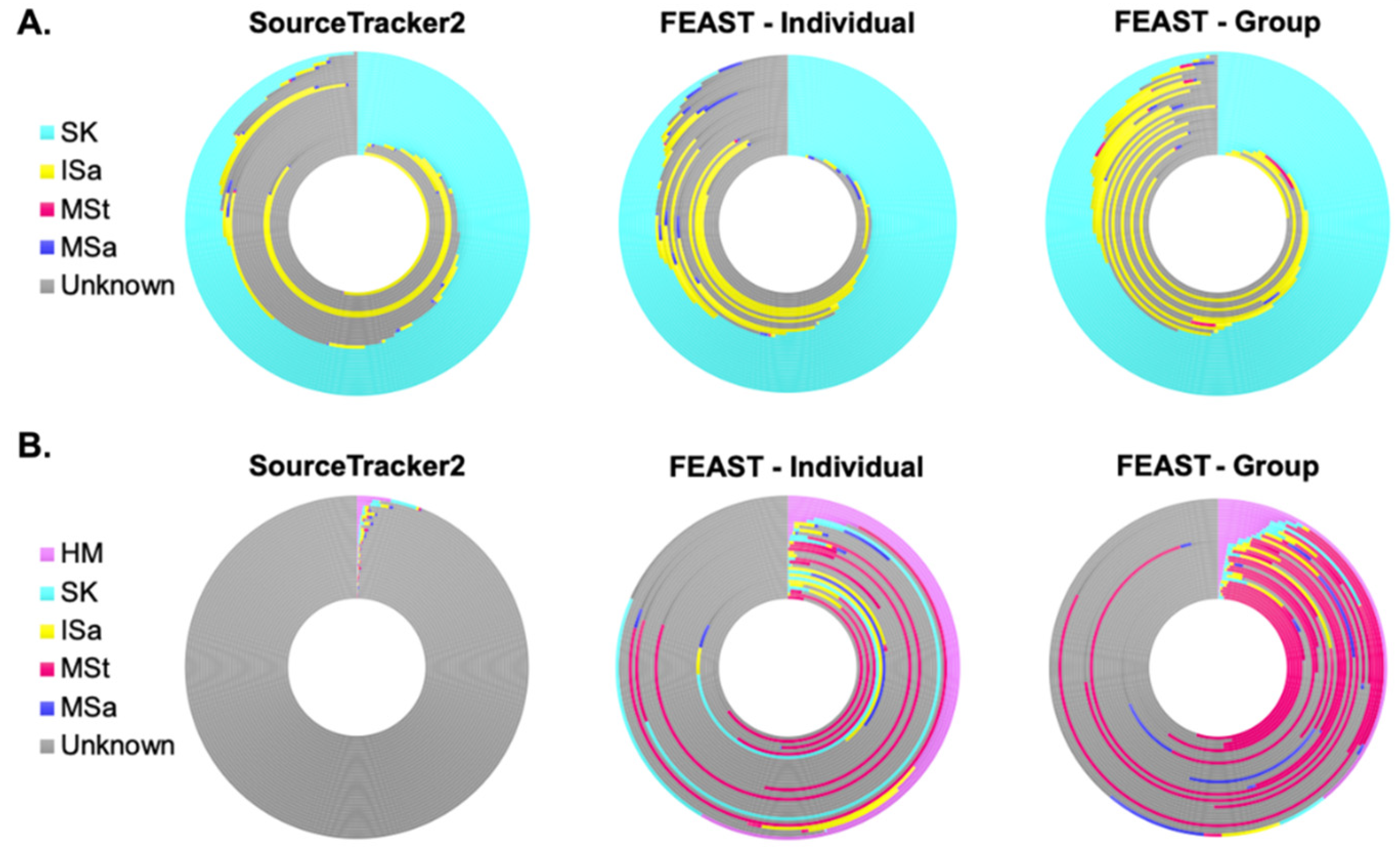
3.5. Microbial Sources Contributing to Human Milk and Infant Fecal Microbiota
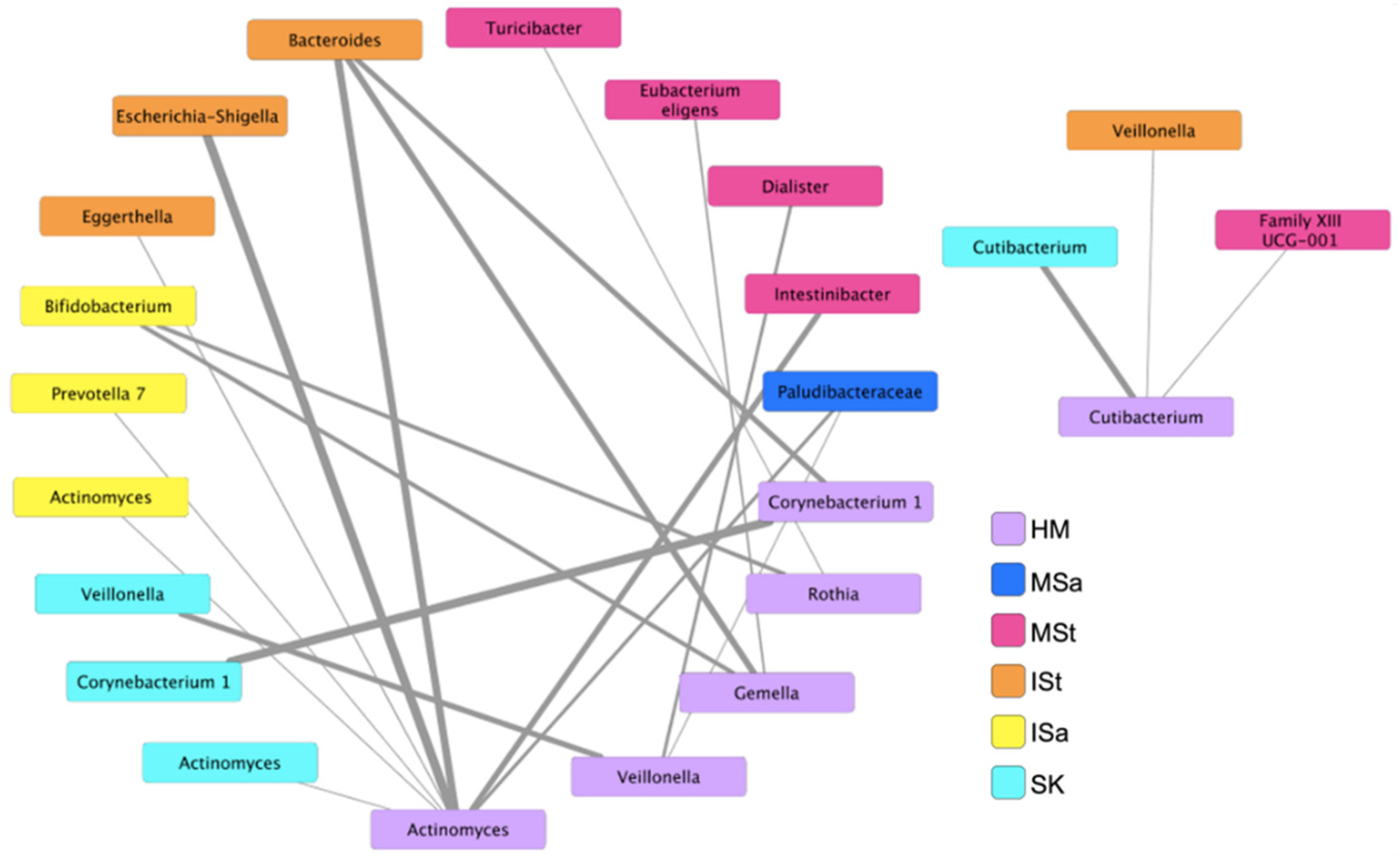
3.6. Bacterial Co-Occurrence Patterns between Human Milk and Maternal and Infant Body Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Monaco, M.H.; Donovan, S.M. Impact of early gut microbiota on immune and metabolic development and function. Semin. Fetal Neonatal. Med. 2016, 21, 380–387. [Google Scholar] [CrossRef]
- Collado, M.C.; Cernada, M.; Bauerl, C.; Vento, M.; Perez-Martinez, G. Microbial ecology and host-microbiota interactions during early life stages. Gut Microbes 2012, 3, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, C.L.; Onnerfalt, J.; Xu, J.; Molin, G.; Ahrne, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obes. (Silver Spring) 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Aomatsu, T.; Imaeda, H.; Fujimoto, T.; Takahashi, K.; Yoden, A.; Tamai, H.; Fujiyama, Y.; Andoh, A. Terminal restriction fragment length polymorphism analysis of the gut microbiota profiles of pediatric patients with inflammatory bowel disease. Digestion 2012, 86, 129–135. [Google Scholar] [CrossRef]
- Ling, Z.; Li, Z.; Liu, X.; Cheng, Y.; Luo, Y.; Tong, X.; Yuan, L.; Wang, Y.; Sun, J.; Li, L.; et al. Altered fecal microbiota composition associated with food allergy in infants. Appl. Env. Microbiol. 2014, 80, 2546–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, M.C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B.; Gold, M.J.; Britton, H.M.; Lefebvre, D.L.; et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.C.; Wang, M.; Donovan, S.M. The role of early life nutrition in the establishment of gastrointestinal microbial composition and function. Gut Microbes 2017, 8, 143–171. [Google Scholar] [CrossRef]
- Moore, R.E.; Townsend, S.D. Temporal development of the infant gut microbiome. Open Biol 2019, 9, 190128. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Hunt, K.M.; Foster, J.A.; Forney, L.J.; Schutte, U.M.; Beck, D.L.; Abdo, Z.; Fox, L.K.; Williams, J.E.; McGuire, M.K.; McGuire, M.A. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 2011, 6, e21313. [Google Scholar] [CrossRef] [Green Version]
- McGuire, M.K.; McGuire, M.A. Human milk: Mother nature’s prototypical probiotic food? Adv. Nutr. 2015, 6, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Boix-Amoros, A.; Collado, M.C.; Mira, A. Relationship between Milk Microbiota, Bacterial Load, Macronutrients, and Human Cells during Lactation. Front. Microbiol. 2016, 7, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera-Rubio, R.; Collado, M.C.; Laitinen, K.; Salminen, S.; Isolauri, E.; Mira, A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 2012, 96, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; du Toit, E.; Kulkarni, A.; Aakko, J.; Linderborg, K.M.; Zhang, Y.; Nicol, M.P.; Isolauri, E.; Yang, B.; Collado, M.C.; et al. Distinct Patterns in Human Milk Microbiota and Fatty Acid Profiles Across Specific Geographic Locations. Front. Microbiol. 2016, 7, 1619. [Google Scholar] [CrossRef] [Green Version]
- Soto, A.; Martin, V.; Jimenez, E.; Mader, I.; Rodriguez, J.M.; Fernandez, L. Lactobacilli and bifidobacteria in human breast milk: Influence of antibiotherapy and other host and clinical factors. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moossavi, S.; Sepehri, S.; Robertson, B.; Bode, L.; Goruk, S.; Field, C.J.; Lix, L.M.; de Souza, R.J.; Becker, A.B.; Mandhane, P.J.; et al. Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 2019, 25, 324–335.e4. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, L.; Langa, S.; Martin, V.; Maldonado, A.; Jimenez, E.; Martin, R.; Rodriguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharm. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M. The origin of human milk bacteria: Is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Lackey, K.A.; Williams, J.E.; Meehan, C.L.; Zachek, J.A.; Benda, E.D.; Price, W.J.; Foster, J.A.; Sellen, D.W.; Kamau-Mbuthia, E.W.; Kamundia, E.W.; et al. What’s Normal? Microbiomes in Human Milk and Infant Feces Are Related to Each Other but Vary Geographically: The INSPIRE Study. Front. Nutr. 2019, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Reyes, S.M.; Allen, D.L.; Williams, J.E.; McGuire, M.A.; McGuire, M.K.; Hay, A.G.; Rasmussen, K.M. Pumping supplies alter the microbiome of pumped human milk: An in-home, randomized, crossover trial. Am. J. Clin. Nutr. 2021, 114, 1960–1970. [Google Scholar] [CrossRef]
- Ho, N.T.; Li, F.; Lee-Sarwar, K.A.; Tun, H.M.; Brown, B.P.; Pannaraj, P.S.; Bender, J.M.; Azad, M.B.; Thompson, A.L.; Weiss, S.T.; et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. 2018, 9, 4169. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 2018, 24, 133–145.e5. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Langa, S.; Reviriego, C.; Jiminez, E.; Marin, M.L.; Xaus, J.; Fernandez, L.; Rodriguez, J.M. Human milk is a source of lactic acid bacteria for the infant gut. J. Pediatr. 2003, 143, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Perez, P.F.; Dore, J.; Leclerc, M.; Levenez, F.; Benyacoub, J.; Serrant, P.; Segura-Roggero, I.; Schiffrin, E.J.; Donnet-Hughes, A. Bacterial imprinting of the neonatal immune system: Lessons from maternal cells? Pediatrics 2007, 119, e724–e732. [Google Scholar] [CrossRef] [PubMed]
- Treven, P.; Mrak, V.; Bogovic Matijasic, B.; Horvat, S.; Rogelj, I. Administration of probiotics Lactobacillus rhamnosus GG and Lactobacillus gasseri K7 during pregnancy and lactation changes mouse mesenteric lymph nodes and mammary gland microbiota. J. Dairy Sci. 2015, 98, 2114–2128. [Google Scholar] [CrossRef] [Green Version]
- De Moreno de LeBlanc, A.; Dogi, C.A.; Galdeano, C.M.; Carmuega, E.; Weill, R.; Perdigon, G. Effect of the administration of a fermented milk containing Lactobacillus casei DN-114001 on intestinal microbiota and gut associated immune cells of nursing mice and after weaning until immune maturity. BMC Immunol. 2008, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Mancabelli, L.; Lugli, G.A.; Duranti, S.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Ferretti, P.; Gorfer, V.; et al. Exploring Vertical Transmission of Bifidobacteria from Mother to Child. Appl Env. Microbiol. 2015, 81, 7078–7087. [Google Scholar] [CrossRef] [Green Version]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Rochat, F.; Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Env. Microbiol. 2014, 16, 2891–2904. [Google Scholar] [CrossRef]
- Drell, T.; Stsepetova, J.; Simm, J.; Rull, K.; Aleksejeva, A.; Antson, A.; Tillmann, V.; Metsis, M.; Sepp, E.; Salumets, A.; et al. The Influence of Different Maternal Microbial Communities on the Development of Infant Gut and Oral Microbiota. Sci. Rep. 2017, 7, 9940. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.E.; Carrothers, J.M.; Lackey, K.A.; Beatty, N.F.; Brooker, S.L.; Peterson, H.K.; Steinkamp, K.M.; York, M.A.; Shafii, B.; Price, W.J.; et al. Strong Multivariate Relations Exist Among Milk, Oral, and Fecal Microbiomes in Mother-Infant Dyads during the First Six Months Postpartum. J. Nutr. 2019, 149, 902–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, K.; Moossavi, S.; Sbihi, H.; Boutin, R.C.T.; Bode, L.; Robertson, B.; Yonemitsu, C.; Field, C.J.; Becker, A.B.; Mandhane, P.J.; et al. Breastmilk Feeding Practices Are Associated with the Co-Occurrence of Bacteria in Mothers’ Milk and the Infant Gut: The CHILD Cohort Study. Cell Host Microbe 2020, 28, 285–297.e4. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef]
- Biagi, E.; Quercia, S.; Aceti, A.; Beghetti, I.; Rampelli, S.; Turroni, S.; Faldella, G.; Candela, M.; Brigidi, P.; Corvaglia, L. The Bacterial Ecosystem of Mother’s Milk and Infant’s Mouth and Gut. Front. Microbiol. 2017, 8, 1214. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.; Curley, D.; O’Callaghan, T.F.; O’Shea, C.A.; Dempsey, E.M.; O’Toole, P.W.; Ross, R.P.; Ryan, C.A.; Stanton, C. The Composition of Human Milk and Infant Faecal Microbiota Over the First Three Months of Life: A Pilot Study. Sci. Rep. 2017, 7, 40597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, D.T.; Kent, J.C.; Owens, R.A.; Hartmann, P.E. Ultrasound imaging of milk ejection in the breast of lactating women. Pediatrics 2004, 113, 361–367. [Google Scholar] [CrossRef]
- Holgerson, P.L.; Vestman, N.R.; Claesson, R.; Ohman, C.; Domellof, M.; Tanner, A.C.; Hernell, O.; Johansson, I. Oral microbial profile discriminates breast-fed from formula-fed infants. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oba, P.M.; Holscher, H.D.; Mathai, R.A.; Kim, J.; Swanson, K.S. Diet Influences the Oral Microbiota of Infants during the First Six Months of Life. Nutrients 2020, 12, 3400. [Google Scholar] [CrossRef]
- LeMay-Nedjelski, L.; Asbury, M.R.; Butcher, J.; Ley, S.H.; Hanley, A.J.; Kiss, A.; Unger, S.; Copeland, J.K.; Wang, P.W.; Stintzi, A.; et al. Maternal Diet and Infant Feeding Practices Are Associated with Variation in the Human Milk Microbiota at 3 Months Postpartum in a Cohort of Women with High Rates of Gestational Glucose Intolerance. J. Nutr. 2021, 151, 320–329. [Google Scholar] [CrossRef]
- Fiese, B.H.; Musaad, S.; Bost, K.K.; McBride, B.A.; Lee, S.Y.; Teran-Garcia, M.; Donovan, S.M. The STRONG Kids 2 Birth Cohort Study: A Cell-to-Society Approach to Dietary Habits and Weight Trajectories across the First 5 Years of Life. Curr. Dev. Nutr. 2019, 3, nzz007. [Google Scholar] [CrossRef]
- Witt, A.; Mason, M.J.; Burgess, K.; Flocke, S.; Zyzanski, S. A case control study of bacterial species and colony count in milk of breastfeeding women with chronic pain. Breastfeed. Med. 2014, 9, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Bauer, L.L.; Chen, X.; Wang, M.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Fahey, G.C.; Donovan, S.M. Microbial composition and in vitro fermentation patterns of human milk oligosaccharides and prebiotics differ between formula-fed and sow-reared piglets. J. Nutr. 2012, 142, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.E.; Carrothers, J.M.; Lackey, K.A.; Beatty, N.F.; York, M.A.; Brooker, S.L.; Shafii, B.; Price, W.J.; Settles, M.L.; McGuire, M.A.; et al. Human Milk Microbial Community Structure Is Relatively Stable and Related to Variations in Macronutrient and Micronutrient Intakes in Healthy Lactating Women. J. Nutr. 2017, 147, 1739–1748. [Google Scholar] [CrossRef]
- Monaco, M.H.; Wang, M.; Pan, X.; Li, Q.; Richards, J.D.; Chichlowski, M.; Berg, B.M.; Dilger, R.N.; Donovan, S.M. Evaluation of Sialyllactose Supplementation of a Prebiotic-Containing Formula on Growth, Intestinal Development, and Bacterial Colonization in the Neonatal Piglet. Curr. Dev. Nutr. 2018, 2, nzy067. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Muturi, E.J.; Donthu, R.K.; Fields, C.J.; Moise, I.K.; Kim, C.H. Effect of pesticides on microbial communities in container aquatic habitats. Sci. Rep. 2017, 7, 44565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Env. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Shenhav, L.; Thompson, M.; Joseph, T.A.; Briscoe, L.; Furman, O.; Bogumil, D.; Mizrahi, I.; Pe’er, I.; Halperin, E. FEAST: Fast expectation-maximization for microbial source tracking. Nat. Methods 2019, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [Green Version]
- Demmelmair, H.; Jimenez, E.; Collado, M.C.; Salminen, S.; McGuire, M.K. Maternal and Perinatal Factors Associated with the Human Milk Microbiome. Curr. Dev. Nutr. 2020, 4, nzaa027. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | EBF (n = 20) | MF (n = 13) |
|---|---|---|
| Maternal age (years) | 32.1 (5.3) | 33.0 (3.8) |
| Maternal BMI (kg/m2) | ||
| Pre-pregnancy | 22.4 (3.0) a | 27.4 (7.9) b |
| 6-weeks postpartum | 24.5 (4.2) | 26.1 (14.7) |
| Delivery mode | ||
| Vaginal delivery | 18 (90) a | 6 (46) b |
| Planned C-section | 1 (5) | 2 (15) |
| Emergency C-section | 1 (5) | 5 (38) |
| Infant gender | ||
| Male | 9 (45) | 9 (69) |
| Female | 11 (55) | 4 (31) |
| Percent feedings from HM | 100 | 67.0 (35.0) |
| Percent feedings from formula | 0 | 33.0 (35.0) |
| Consumed prenatal probiotics | 3 (15) | 0 |
| Regular pumping at 6-weeks | ||
| Yes | 9 (45) | 7 (58) |
| No | 9 (45) | 5 (42) |
| Unknown | 2 (10) | 1 (3) |
| Shannon Index | Observed Features | Faith Phylogenetic Diversity | |
|---|---|---|---|
| Human milk | |||
| EBF (n = 20) | 2.3 ± 0.2 | 22.9 ± 1.9 | 18.2 ± 1.5 |
| MF (n = 13) | 2.6 ± 0.3 | 29.2 ± 4.0 | 14.3 ± 1.8 |
| Breast skin | |||
| EBF (n = 20) | 3.0 ± 0.1 a | 41.9 ± 3.9 | 10.2 ± 0.4 |
| MF (n = 13) | 3.5 ± 0.3 b | 52.0 ± 6.7 | 10.1 ± 0.5 |
| Infant saliva | |||
| EBF (n = 20) | 2.1 ± 0.2 a | 25.4 ± 1.3 a | 13.8 ± 1.0 |
| MF (n = 13) | 3.1 ± 0.2 b | 36.7 ± 3.8 b | 13.6 ± 1.3 |
| Maternal saliva | |||
| EBF (n = 18) | 5.3 ± 0.1 | 152.8 ± 5.6 | 14.7 ± 0.4 |
| MF (n = 13) | 5.3 ± 0.1 | 160.6 ± 9.5 | 15.5 ± 1.0 |
| Infant stool | |||
| EBF (n = 20) | 2.6 ± 0.2 | 22.8 ± 1.5 | 6.9 ± 0.1 |
| MF (n = 12) | 2.5 ± 0.2 | 24.7 ± 1.8 | 7.3 ± 0.2 |
| Maternal stool | |||
| EBF (n = 18) | 5.2 ± 0.1 | 101.2 ± 4.9 | 12.4 ± 0.3 |
| MF (n = 13) | 4.9 ± 0.2 | 94.2 ± 6.9 | 11.9 ± 0.5 |
| Genus | Human Milk | Breast Skin | Infant Saliva | Infant Stool | Maternal Saliva | Maternal Stool | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EBF | MF | EBF | MF | EBF | MF | EBF | MF | EBF | MF | EBF | MF | |
| Staphylococcus | 45.4 ± 6.8 | 48.8 ± 9.9 | 32.7 ± 5.3 | 37.5 ± 8.2 | 0.4 ± 0.2 | 1.0 ± 0.8 | ||||||
| Streptococcus | 33.4 ± 5.9 † | 19.0 ± 5.9 | 41.7 ± 4.0 | 31.4 ± 5.1 | 67.4 ± 3.5 * | 53.5 ± 4.5 | 2.1 ± 0.6 | 2.3 ± 1.1 | 17.3 ± 1.5 | 19.3 ± 2.1 | ||
| Corynebacterium 1 | 4.1 ± 1.8 | 2.9 ± 1.2 | 7.1 ± 1.6 | 5.4 ± 1.2 | ||||||||
| Rothia | 3.5 ± 1.6 | 1.3 ± 0.4 | 2.0 ± 0.5 | 2.6 ± 1.2 | 3.6 ± 1.3 | 9.6 ± 3.1 | 2.9 ± 0.6 | 3.2 ± 0.7 | ||||
| Gemella | 1.9 ± 1.0 | 0.6 ± 0.4 | 3.0 ± 0.9 | 1.5 ± 0.6 | 15.1 ± 2.3 * | 7.9 ± 2.0 | ||||||
| Veillonella | 1.1 ± 0.7 | 1.8 ± 1.4 | 2.0 ± 0.5 | 3.1 ± 0.8 | 3.9 ± 1.2 * | 10.7 ± 2.1 | 4.0 ± 1.9 | 4.7 ± 3.7 | 6.3 ± 0.7 | 6.0 ± 1.1 | ||
| Cutibacterium | 1.3 ± 0.7 | 0.7 ± 0.4 | ||||||||||
| Bifidobacterium | 0.2 ± 0.2 | 0.6 ± 0.6 | 17.1 ± 4.7 | 31.2 ± 6.7 | 3.1 ± 1.3 | 2.1 ± 0.7 | ||||||
| Actinomyces | 0.2 ± 0.1 | 0.3 ± 0.2 | 1.0 ± 0.4 | 1.1 ± 0.5 | ||||||||
| Streptomyces | 0.3 ± 0.1 | 0.1 ± 0.1 | ||||||||||
| Lactobacillus | 2.0 ± 1.9 | 0.4 ± 0.2 | ||||||||||
| Prevotella 7 | 0.4 ± 0.4 | 1.9 ± 1.3 | 0.4 ± 0.3 | 2.5 ± 1.8 | 17.1 ± 1.7 | 13.5 ± 1.4 | ||||||
| Haemophilus | 0.6 ± 0.4 | 1.5 ± 1.1 | 5.2 ± 2.4 | 2.9 ± 2.6 | 10.0 ± 1.3 | 7.7 ± 1.2 | ||||||
| Atopobium | 1.0 ± 0.8 | 1.3 ± 1.0 | ||||||||||
| Neisseria | 0.0 ± 0.0 | 2.4 ± 2.4 | 8.7 ± 1.9 | 11.7 ± 2.3 | ||||||||
| Porphyromonas | 0.5 ± 0.5 | 0.8 ± 0.5 | 3.3 ± 0.9 | 3.4 ± 0.8 | ||||||||
| Escherichia-Shigella | 16.0 ± 5.0 | 17.1 ± 6.3 | 2.1 ± 2.1 | 4.5 ± 3.1 | ||||||||
| Bacteroides | 17.0 ± 5.1 | 7.0 ± 4.9 | 17.0 ± 2.2 † | 18.7 ± 3.9 | ||||||||
| Klebsiella | 9.4 ± 3.9 | 6.8 ± 4.0 | ||||||||||
| Clostridium sensu stricto 1 | 7.2 ± 3.1 | 0.8 ± 0.5 | ||||||||||
| Enterobacter | 6.2 ± 3.6 | 2.1 ± 1.5 | ||||||||||
| Enterococcus | 2.0 ± 0.9 | 5.3 ± 3.7 | ||||||||||
| Ruminococcus gnavus group | 0.6 ± 0.4 | 4.0 ± 2.6 | ||||||||||
| Fusobacterium | 6.4 ± 1.2 * | 10.0 ± 1.2 | ||||||||||
| Alloprevotella | 5.0 ± 0.5 | 4.1 ± 0.8 | ||||||||||
| Leptotrichia | 2.7 ± 0.5 | 3.1 ± 1.4 | ||||||||||
| Faecalibacterium | 12.2 ± 2.1 | 9.9 ± 2.4 | ||||||||||
| Blautia | 7.2 ± 1.1 | 9.8 ± 2.3 | ||||||||||
| Akkermansia | 4.8 ± 1.6 | 5.3 ± 2.2 | ||||||||||
| Subdoligranulum | 3.5 ± 1.2 | 3.4 ± 1.0 | ||||||||||
| Eubacterium hallii group | 3.6 ± 0.6 | 3.1 ± 0.9 | ||||||||||
| Ruminococcus 2 | 3.5 ± 1.1 | 2.7 ± 0.9 | ||||||||||
| Dialister | 1.1 ± 0.6 | 4.4 ± 2.6 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, E.C.; Wang, M.; Donovan, S.M. Microbial Interrelationships across Sites of Breastfeeding Mothers and Infants at 6 Weeks Postpartum. Microorganisms 2022, 10, 1155. https://doi.org/10.3390/microorganisms10061155
Davis EC, Wang M, Donovan SM. Microbial Interrelationships across Sites of Breastfeeding Mothers and Infants at 6 Weeks Postpartum. Microorganisms. 2022; 10(6):1155. https://doi.org/10.3390/microorganisms10061155
Chicago/Turabian StyleDavis, Erin C., Mei Wang, and Sharon M. Donovan. 2022. "Microbial Interrelationships across Sites of Breastfeeding Mothers and Infants at 6 Weeks Postpartum" Microorganisms 10, no. 6: 1155. https://doi.org/10.3390/microorganisms10061155
APA StyleDavis, E. C., Wang, M., & Donovan, S. M. (2022). Microbial Interrelationships across Sites of Breastfeeding Mothers and Infants at 6 Weeks Postpartum. Microorganisms, 10(6), 1155. https://doi.org/10.3390/microorganisms10061155