
Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioethical Requirements
2.2. Sputum Collection and Processing
2.3. Antibacterial Susceptibility
2.4. Whole-Genome Sequencing
2.5. Genomic Analysis
2.6. GWAS
2.7. Serial Isolates Comparisons
3. Results
3.1. Population Structure, Phylogeny, and Drug Susceptibility Profiling
3.1.1. Strains Classification
3.1.2. Drug Resistance Profiling
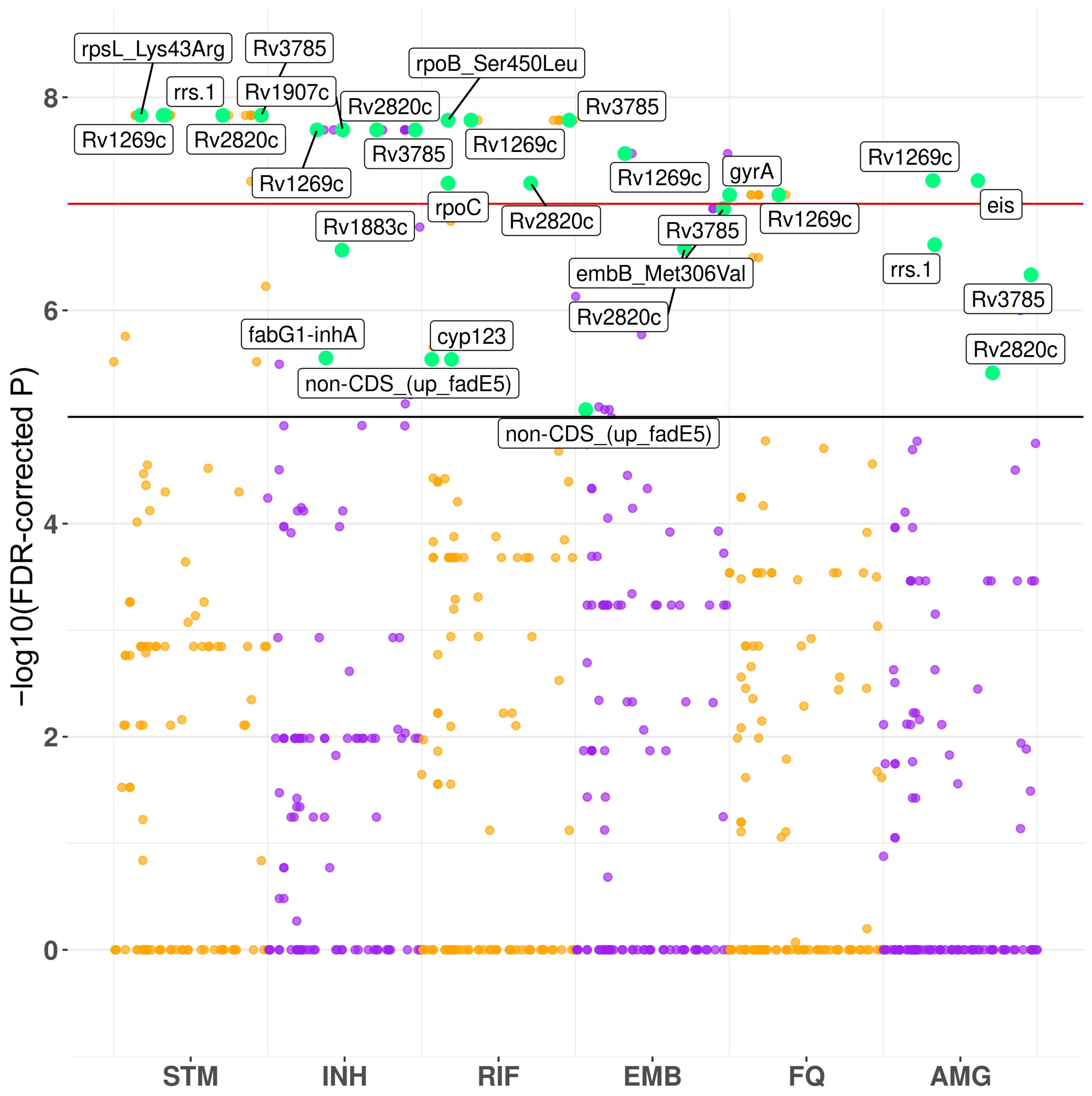
3.2. Convergence-Based Search of Resistance-Associated Polymorphisms
3.3. Serial Isolates Comparisons
4. Discussion
4.1. GWAS Hits
4.2. SNPs and Indels Emerged during the Treatment
4.3. Insertion of 1 bp within Rv3785 Was Associated with Drug Resistance and Emerged after Anti-TB Treatment in Beijing Isolates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240037021.
- World Health Organization. WHO Global Lists of High Burden Countries for Tuberculosis (TB), TB/HIV and TB (MDR/RR-TB); World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240029439.
- Ozaki, K.; Ohnishi, Y.; Iida, A.; Sekine, A.; Yamada, R.; Tsunoda, T.; Sato, H.; Sato, H.; Hori, M.; Nakamura, Y.; et al. Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 2002, 32, 650–654. [Google Scholar] [CrossRef]
- Power, R.A.; Parkhill, J.; De Oliveira, T. Microbial genome-wide association studies: Lessons from human GWAS. Nat. Rev. Genet. 2016, 18, 41–50. [Google Scholar] [CrossRef]
- Chen, P.E.; Shapiro, B.J. The advent of genome-wide association studies for bacteria. Curr. Opin. Microbiol. 2015, 25, 17–24. [Google Scholar] [CrossRef]
- Farhat, M.R.; Shapiro, B.J.; Kieser, K.J.; Sultana, R.; Jacobson, K.R.; Victor, T.C.; Warren, R.M.; Streicher, E.M.; Calver, A.; Sloutsky, A.; et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2013, 45, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Laabei, M.; Recker, M.; Rudkin, J.K.; Aldeljawi, M.; Gulay, Z.; Sloan, T.J.; Williams, P.; Endres, J.L.; Bayles, K.W.; Fey, P.D.; et al. Predicting the virulence of MRSA from its genome sequence. Genome Res. 2014, 24, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.; Wolff, K.A.; Nguyen, L. Molecular biology of drug resistance in Mycobacterium tuberculosis. In Pathogenesis of Mycobacterium tuberculosis and Its Interaction with the Host Organism; Springer: Berlin/Heidelberg, Germany, 2012; pp. 53–80. [Google Scholar]
- Merker, M.; Barbier, M.; Cox, H.; Rasigade, J.P.; Feuerriegel, S.; Kohl, T.A.; Diel, R.; Borrell, S.; Gagneux, S.; Nikolayevskyy, V.; et al. Compensatory evolution drives multidrug-resistant tuberculosis in central Asia. Elife 2018, 7, e18103. [Google Scholar] [CrossRef]
- Müller, B.; Borrell, S.; Rose, G.; Gagneux, S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013, 29, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Datta, G.; Nieto, L.M.; Davidson, R.M.; Mehaffy, C.; Pederson, C.; Dobos, K.M.; Strong, M. Longitudinal whole genome analysis of pre and post drug treatment Mycobacterium tuberculosis isolates reveals progressive steps to drug resistance. Tuberculosis 2016, 98, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Walter, N.D.; Dolganov, G.M.; Garcia, B.J.; Worodria, W.; Andama, A.; Musisi, E.; Ayakaka, I.; Van, T.T.; Voskuil, M.I.; de Jong, B.C.; et al. Transcriptional adaptation of drug-tolerant Mycobacterium tuberculosis during treatment of human tuberculosis. J. Infect. Dis. 2015, 212, 990–998. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Lee, S.K.; Song, N.; Nathan, T.O.; Swarts, B.M.; Eum, S.Y.; Ehrt, S.; Cho, S.N.; Eoh, H. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 2019, 10, 2928. [Google Scholar] [CrossRef]
- Hjort, K.; Jurén, P.; Toro, J.C.; Hoffner, S.; Andersson, D.I.; Sandegren, L. Dynamics of extensive drug resistance evolution of Mycobacterium tuberculosis in a single patient during 9 years of disease and treatment. J. Infect. Dis. 2022, 225, 1011–1020. [Google Scholar] [CrossRef]
- Sun, G.; Luo, T.; Yang, C.; Dong, X.; Li, J.; Zhu, Y.; Zheng, H.; Tian, W.; Wang, S.; Barry, C.E.; et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J. Infect. Dis. 2012, 206, 1724–1733. [Google Scholar] [CrossRef]
- Kato-Maeda, M.; Ho, C.; Passarelli, B.; Banaei, N.; Grinsdale, J.; Flores, L.; Anderson, J.; Murray, M.; Rose, G.; Kawamura, L.M.; et al. Use of whole genome sequencing to determine the microevolution of Mycobacterium tuberculosis during an outbreak. PLoS ONE 2013, 8, e58235. [Google Scholar] [CrossRef] [Green Version]
- Merker, M.; Kohl, T.A.; Roetzer, A.; Truebe, L.; Richter, E.; Rüsch-Gerdes, S.; Fattorini, L.; Oggioni, M.R.; Cox, H.; Varaine, F.; et al. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PLoS ONE 2013, 8, e82551. [Google Scholar] [CrossRef] [Green Version]
- Saunders, N.J.; Trivedi, U.H.; Thomson, M.L.; Doig, C.; Laurenson, I.F.; Blaxter, M.L. Deep resequencing of serial sputum isolates of Mycobacterium tuberculosis during therapeutic failure due to poor compliance reveals stepwise mutation of key resistance genes on an otherwise stable genetic background. J. Infect. 2011, 62, 212–217. [Google Scholar] [CrossRef]
- Fursov, M.V.; Shitikov, E.A.; Lagutkin, D.A.; Fursova, A.D.; Ganina, E.A.; Kombarova, T.I.; Grishenko, N.S.; Rudnitskaya, T.I.; Bespiatykh, D.A.; Kolupaeva, N.V.; et al. MDR and Pre-XDR clinical Mycobacterium tuberculosis beijing strains: Assessment of virulence and host cytokine response in mice infectious model. Microorganisms 2021, 9, 1792. [Google Scholar] [CrossRef]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 June 2022).
- Snippy. Available online: https://github.com/tseemann/snippy (accessed on 1 June 2022).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, btv566. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. genomics 2016, 2, e000056. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (ITOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Kohl, T.A.; Utpatel, C.; Schleusener, V.; De Filippo, M.R.; Beckert, P.; Cirillo, D.M.; Niemann, S. MTBseq: A comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 2018, 2018, e5895. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of drosophila melanogaster strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Saund, K.; Lapp, Z.; Thiede, S.N.; Pirani, A.; Snitkin, E.S. Prewas: Data pre-processing for more informative bacterial gwas. Microb. Genomics 2020, 6, e000368. [Google Scholar] [CrossRef]
- Saund, K.; Snitkin, E.S. Hogwash: Three methods for genome-wide association studies in bacteria. Microb. Genomics 2020, 6, mgen000469. [Google Scholar] [CrossRef]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A hidden markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [Green Version]
- Ajawatanawong, P.; Yanai, H.; Smittipat, N.; Disratthakit, A.; Yamada, N.; Miyahara, R.; Nedsuwan, S.; Imasanguan, W.; Kantipong, P.; Chaiyasirinroje, B.; et al. A novel ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to modern beijing sublineages. Sci. Rep. 2019, 9, 13718. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; deVisch, A.; Barthe, P.; Turapov, O.; Aydogan, T.; Heriaud, L.; Gracy, J.; Mukamolova, G.V.; Letourneur, F.; Cohen-Gonsaud, M. A Mycobacterium tuberculosis effector targets mitochondrion, controls energy metabolism and limits cytochrome c exit. bioRxiv 2021. [Google Scholar] [CrossRef]
- De Keijzer, J.; De Haas, P.E.; De Ru, A.H.; Van Veelen, P.A.; Van Soolingen, D. Disclosure of selective advantages in the “modern” sublineage of the Mycobacterium tuberculosis beijing genotype family by quantitative proteomics. Mol. Cell. Proteom. 2014, 13, 2632–2645. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, R.K.; Gößringer, M.; Späth, B.; Fischer, S.; Marchfelder, A. Chapter 8 The making of TRNAs and more—RNase P and TRNase Z. In Progress in Molecular Biology and Translational Science; Academic Press: Cambridge, MA, USA, 2009; Volume 85, pp. 319–368. [Google Scholar]
- Carlson, B.L.; Ballister, E.R.; Skordalakes, E.; King, D.S.; Breidenbach, M.A.; Gilmore, S.A.; Berger, J.M.; Bertozzi, C.R. Function and structure of a prokaryotic formylglycine-generating enzyme. J. Biol. Chem. 2008, 283, 20117–20125. [Google Scholar] [CrossRef] [Green Version]
- Beatty, K.E.; Williams, M.; Carlson, B.L.; Swarts, B.M.; Warren, R.M.; Van Helden, P.D.; Bertozzi, C.R. Sulfatase-activated fluorophores for rapid discrimination of mycobacterial species and strains. Proc. Natl. Acad. Sci. USA 2013, 110, 12911–12916. [Google Scholar] [CrossRef] [Green Version]
- Sogi, K.M.; Gartner, Z.J.; Breidenbach, M.A.; Appel, M.J.; Schelle, M.W.; Bertozzi, C.R. Mycobacterium tuberculosis Rv3406 is a type II alkyl sulfatase capable of sulfate scavenging. PLoS ONE 2013, 8, e65080. [Google Scholar] [CrossRef]
- Akhter, Y.; Yellaboina, S.; Farhana, A.; Ranjan, A.; Ahmed, N.; Hasnain, S.E. Genome scale portrait of CAMP-receptor protein (CRP) regulons in mycobacteria points to their role in pathogenesis. Gene 2008, 407, 148–158. [Google Scholar] [CrossRef]
- Zhang, Q.; Wan, B.; Zhou, A.; Ni, J.; Xu, Z.; Li, S.; Tao, J.; Yao, Y.F. Whole genome analysis of an MDR Beijing/W strain of Mycobacterium tuberculosis with large genomic deletions associated with resistance to isoniazid. Gene 2016, 582, 128–136. [Google Scholar] [CrossRef]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE 2010, 5, e13938. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chen, J.; Yan, B.; Zhang, W.; Guddat, L.W.; Liu, X.; Rao, Z. Structural basis for the broad substrate specificity of two Acyl-CoA dehydrogenases FadE5 from mycobacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 16324–16332. [Google Scholar] [CrossRef]
- Calamita, H.; Ko, C.; Tyagi, S.; Yoshimatsu, T.; Morrison, N.E.; Bishai, W.R. The Mycobacterium tuberculosis SigD sigma factor controls the expression of ribosome-associated gene products in stationary phase and is required for full virulence. Cell. Microbiol. 2004, 7, 233–244. [Google Scholar] [CrossRef]
- Respicio, L.; Nair, P.A.; Huang, Q.; Anil, B.; Tracz, S.; Truglio, J.J.; Kisker, C.; Raleigh, D.P.; Ojima, I.; Knudson, D.L.; et al. Characterizing septum inhibition in Mycobacterium tuberculosis for novel drug discovery. Tuberculosis 2008, 88, 420–429. [Google Scholar] [CrossRef]
- Kandler, J.L.; Mercante, A.D.; Dalton, T.L.; Ezewudo, M.N.; Cowan, L.S.; Burns, S.P.; Metchock, B.; Cegielski, P.; Posey, J.E. Validation of novel Mycobacterium tuberculosis isoniazid resistance mutations not detectable by common molecular tests. Antimicrob. Agents Chemother. 2018, 62, e00974-18. [Google Scholar] [CrossRef] [Green Version]
- Voskuil, M.I.; Bartek, I.L.; Visconti, K.; Schoolnik, G.K. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2011, 2, 105. [Google Scholar] [CrossRef] [Green Version]
- Raman, K.; Chandra, N.M. Tuberculosis interactome analysis unravels potential pathways to drug resistance. Nat. Preced. 2008, 1. [Google Scholar] [CrossRef]
- Stavrum, R.; Valvatne, H.; Bø, T.H.; Jonassen, I.; Hinds, J.; Butcher, P.D.; Grewal, H.M.S. Genomic diversity among Beijing and non-Beijing Mycobacterium tuberculosis isolates from myanmar. PLoS ONE 2008, 3, e1973. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Zhang, S.; Fleming, J.; Chen, Y.; Li, Z.; Fan, S.; Liu, Y.; Wang, W.; Wang, T.; Liu, Y.; et al. Mycobacterium tuberculosis type III-A CRISPR/Cas system CrRNA and its maturation have atypical features. FASEB J. 2019, 33, 1496–1509. [Google Scholar] [CrossRef]
- Zhai, X.; Luo, T.; Peng, X.; Ma, P.; Wang, C.; Zhang, C.; Suo, J.; Bao, L. The truncated Rv2820c of Mycobacterium tuberculosis Beijing family augments intracellular survival of M. smegmatis by altering cytokine profile and inhibiting NO generation. Infect. Genet. Evol. 2018, 59, 75–83. [Google Scholar] [CrossRef]
- Lam, J.T.; Yuen, K.Y.; Ho, P.L.; Weng, X.H.; Zhang, W.H.; Chen, S.; Yam, W.C. Truncated Rv2820c enhances mycobacterial virulence ex vivo and in vivo. Microb. Pathog. 2011, 50, 331–335. [Google Scholar] [CrossRef]
- Coll, F.; Phelan, J.; Hill-Cawthorne, G.A.; Nair, M.B.; Mallard, K.; Ali, S.; Abdallah, A.M.; Alghamdi, S.; Alsomali, M.; Ahmed, A.O.; et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2018, 50, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Singhal, N.; Sharma, P.; Kumar, M.; Joshi, B.; Bisht, D. Analysis of intracellular expressed proteins of Mycobacterium tuberculosis clinical isolates. Proteome Sci. 2012, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Dubnau, E.; Fontán, P.; Manganelli, R.; Soares-Appel, S.; Smith, I. Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect. Immun. 2002, 70, 2787–2795. [Google Scholar] [CrossRef] [Green Version]
- Dubnau, E.; Chan, J.; Mohan, V.P.; Smith, I. Responses of Mycobacterium tuberculosis to growth in the mouse lung. Infect. Immun. 2005, 73, 3754–3757. [Google Scholar] [CrossRef] [Green Version]
- Ansong, C.; Ortega, C.; Payne, S.H.; Haft, D.H.; Chauvignè-Hines, L.M.; Lewis, M.P.; Ollodart, A.R.; Purvine, S.O.; Shukla, A.K.; Fortuin, S.; et al. Identification of widespread adenosine nucleotide binding in Mycobacterium tuberculosis. Chem. Biol. 2013, 20, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Modlin, S.J.; Elghraoui, A.; Gunasekaran, D.; Zlotnicki, A.M.; Dillon, N.A.; Dhillon, N.; Kuo, N.; Robinhold, C.; Chan, C.K.; Baughn, A.D.; et al. Structure-aware Mycobacterium tuberculosis functional annotation uncloaks resistance, metabolic, and virulence genes. mSystems 2021, 6, e00673-21. [Google Scholar] [CrossRef]
- Wagner, J.; Gruz, P.; Kim, S.R.; Yamada, M.; Matsui, K.; Fuchs, R.P.P.; Nohmi, T. The DinB gene encodes a novel E. Coli DNA polymerase, DNA Pol IV, involved in mutagenesis. Mol. Cell 1999, 4, 281–286. [Google Scholar] [CrossRef]
- Gandotra, S.; Lebron, M.B.; Ehrt, S. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 2010, 6, e1001040. [Google Scholar] [CrossRef]
- Prathiviraj, R.; Chellapandi, P. Deciphering molecular virulence mechanism of Mycobacterium tuberculosis dop isopeptidase based on its sequence–structure–function linkage. Protein J. 2020, 39, 33–45. [Google Scholar] [CrossRef]
- Muzondiwa, D.; Hlanze, H.; Reva, O.N. The epistatic landscape of antibiotic resistance of different clades of Mycobacterium tuberculosis. Antibiotics 2021, 10, 857. [Google Scholar] [CrossRef]
- Bisson, G.P.; Mehaffy, C.; Broeckling, C.; Prenni, J.; Rifat, D.; Lun, D.S.; Burgos, M.; Weissman, D.; Petros, C.K.; Dobosc, K. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, RpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 6441–6452. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Tang, X.; Guo, N.; Zhang, K.; Guo, A.; Wu, X.; Wang, X.; Guan, Z.; Liu, L.; Shen, F.; et al. Genome-wide expression profiling of the response to linezolid in Mycobacterium tuberculosis. Curr. Microbiol. 2012, 64, 530–538. [Google Scholar] [CrossRef]
- de Souza, G.A.; Leversen, N.A.; Målen, H.; Wiker, H.G. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics 2011, 75, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Målen, H.; Berven, F.S.; Fladmark, K.E.; Wiker, H.G. Comprehensive analysis of exported proteins fromMycobacterium tuberculosis H37Rv. Proteomics 2007, 7, 1702–1718. [Google Scholar] [CrossRef]
- Saint-Joanis, B.; Demangel, C.; Jackson, M.; Brodin, P.; Marsollier, L.; Boshoff, H.; Cole, S.T. Inactivation of Rv2525c, a substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases β-Lactam susceptibility and virulence. J. Bacteriol. 2006, 188, 6669–6679. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, S.; Cui, P.; Shi, W.; Zhang, W.; Zhang, Y. Identification of novel mutations associated with cycloserine resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2017, 72, 3272–3276. [Google Scholar] [CrossRef] [Green Version]
- Marbaix, A.Y.; Noël, G.; Detroux, A.M.; Vertommen, D.; Van Schaftingen, E.; Linster, C.L. Extremely conserved ATP-or ADP-dependent enzymatic system for nicotinamide nucleotide. J. Biol. Chem. 2011, 286, 41246–41252. [Google Scholar] [CrossRef] [Green Version]
- AlphaFold Nnr Protein Structure. Available online: https://alphafold.ebi.ac.uk/entry/P9WF11 (accessed on 13 June 2022).
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Ruesen, C.; Chaidir, L.; van Laarhoven, A.; Dian, S.; Ganiem, A.R.; Nebenzahl-Guimaraes, H.; Huynen, M.A.; Alisjahbana, B.; Dutilh, B.E.; van Crevel, R. Large-scale genomic analysis shows association between homoplastic genetic variation in Mycobacterium tuberculosis genes and meningeal or pulmonary tuberculosis. BMC Genomics 2018, 19, 122. [Google Scholar] [CrossRef] [Green Version]
- Milano, A.; Pasca, M.R.; Provvedi, R.; Lucarelli, A.P.; Manina, G.; Luisa de Jesus Lopes Ribeiro, A.; Manganelli, R.; Riccardi, G. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis 2009, 89, 84–90. [Google Scholar] [CrossRef]
- Ismail, N.; Peters, R.P.H.; Ismail, N.A.; Omar, S.V. Clofazimine exposure in vitro selects efflux pump mutants and bedaquiline resistance. Antimicrob. Agents Chemother. 2019, 63, e02141-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, J.; Cui, P.; Shi, W.; Zhang, W.; Zhang, Y. Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2015, 70, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, A.; Kumar, N.; Wright, C.C.; Chou, T.H.; Tringides, M.L.; Bolla, J.R.; Lei, H.T.; Rajashankar, K.R.; Su, C.C.; Purdy, G.E.; et al. Crystal structure of the transcriptional regulator Rv0678 of Mycobacterium tuberculosis. J. Biol. Chem. 2014, 289, 16526–16540. [Google Scholar] [CrossRef] [Green Version]
- Saeed, D.K.; Shakoor, S.; Razzak, S.A.; Hasan, Z.; Sabzwari, S.F.; Azizullah, Z.; Kanji, A.; Nasir, A.; Shafiq, S.; Ghanchi, N.K.; et al. Variants associated with bedaquiline (BDQ) resistance identified in Rv0678 and efflux pump genes in Mycobacterium tuberculosis isolates from BDQ naïve TB patients in pakistan. BMC Microbiol. 2022, 22, 62. [Google Scholar] [CrossRef]
- Villellas, C.; Coeck, N.; Meehan, C.J.; Lounis, N.; de Jong, B.; Rigouts, L.; Andries, K. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J. Antimicrob. Chemother. 2016, 72, dkw502. [Google Scholar] [CrossRef] [Green Version]
- Deshayes, C.; Perrodou, E.; Euphrasie, D.; Frapy, E.; Poch, O.; Bifani, P.; Lecompte, O.; Reyrat, J.M. Detecting the molecular scars of evolution in the Mycobacterium tuberculosis complex by analyzing interrupted coding sequences. BMC Evol. Biol. 2008, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Dianišková, P.; Kordulaḱová, J.; Sǩovierová, H.; Kaur, D.; Jackson, M.; Brennan, P.J.; Mikusǒvá, K. Investigation of ABC transporter from mycobacterial arabinogalactan biosynthetic cluster. Gen. Physiol. Biophys. 2011, 30, 239–250. [Google Scholar] [CrossRef]
- Islam, R.; Brown, S.; Taheri, A.; Dumenyo, C.K. The gene encoding NAD-dependent epimerase/dehydratase, WcaG, affects cell surface properties, virulence, and extracellular enzyme production in the soft rot phytopathogen, pectobacterium carotovorum. Microorganisms 2019, 7, 172. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Lineage | Sublineage | Branch Name | Isolates (%all) | |
|---|---|---|---|---|
| 2 | 2.2.2 | Beijing Ancestral 1 | 1 (0.88%) | 85 (74.56%) |
| 2.2.1 | Beijing Ancestral 2 | 2 (1.75%) | ||
| 2.2.1 | Beijing Asian/Africa 2 | 1 (0.88%) | ||
| 2.2.1 | Beijing Central Asia | 41 (35.96%) | ||
| 2.2.1 | Beijing Central Asia Outbreak | 4 (3.51%) | ||
| 2.2.1 | Beijing B0/W148 | 35 (30.7%) | ||
| 2.2.1 | Beijing (unclassified) | 1 (0.88%) | ||
| 4 | 4.1 | Euro-American | 3 (2.63%) | 29 (25.44%) |
| 4.1.2 | Euro-American | 1 (0.88%) | ||
| 4.1.2.1 | Haarlem | 3 (2.63%) | ||
| 4.2.1 | Ural | 5 (4.39%) | ||
| 4.3.3 | LAM | 11 (9.65%) | ||
| 4.8 | Euro-American (T-family) | 6 (5.26%) | ||
| Streptomycin | Isoniazid | Rifampicin | ||||||
| Position | FDR-Corrected p | Gene | Position | FDR-Corrected p | Gene | Position | FDR-Corrected p | Gene |
| 781687 | 1.48 × 10−8 | rpsL | 1418863 | 2.02 × 10−8 | Rv1269c | 761155 | 1.64 × 10−8 | rpoB |
| 1473246 | 1.48 × 10−8 | rrs | 2153725 | 2.02 × 10−8 | Rv1907c | 1418863 | 1.64 × 10−8 | Rv1269c |
| 1418863 | 1.48 × 10−8 | Rv1269c | 3127931 | 2.02 × 10−8 | Rv2820c | 4231948 | 1.64 × 10−8 | Rv3785 |
| 3127931 | 1.48 × 10−8 | Rv2820c | 4231948 | 2.02 × 10−8 | Rv3785 | 764841 | 6.39 × 10−8 | rpoC |
| 4231948 | 1.48 × 10−8 | Rv3785 | 2133468 | 2.72 × 10−7 | Rv1883c | 3127931 | 6.39 × 10−8 | Rv2820c |
| 2704884 | 3.04 × 10−5 | Rv2407 | 1673425 | 2.79 × 10−6 | fabG1-inhA promoter | 295719 | 2.88 × 10−6 | non-CDS |
| 859498 | 3.42 × 10−5 | cyp123 | 2704884 | 1.20 × 10−5 | Rv2407 | 859498 | 2.88 × 10−6 | cyp123 |
| 859498 | 7.58 × 10−5 | cyp123 | ||||||
| 2155168 | 7.58 × 10−5 | katG | ||||||
| Ethambutol | Fluoroquinolones | Aminoglycosides | ||||||
| Position | FDR-Corrected p | Gene | Position | FDR-Corrected p | Gene | Position | FDR-Corrected p | Gene |
| 1418863 | 3.37 × 10−8 | Rv1269c | 7582 | 8.25 × 10−8 | gyrA | 2715342 | 6.05 × 10−8 | eis promoter |
| 4247429 | 1.11 × 10−7 | embB | 1418863 | 8.25 × 10−8 | Rv1269c | 1418863 | 6.05 × 10−8 | Rv1269c |
| 3127931 | 2.62 × 10−7 | Rv2820c | 7570 | 5.33 × 10−7 | gyrA | 1473246 | 2.42 × 10−7 | rrs |
| 295719 | 8.55 × 10−6 | non-CDS | 2704884 | 1.97 × 10−5 | Rv2407 | 4231948 | 4.64 × 10−7 | Rv3785 |
| 3127931 | 3.86 × 10−6 | Rv2820c | ||||||
| ID | Treatment | Genotype Transitions | ||||||
|---|---|---|---|---|---|---|---|---|
| Rv1435c | Rv0036c | Rv0678 | Rv3433c | dop | ppsA | Rv3785 * | ||
| 144-3289 | Fq Z Cs Lzd Bdq | Cys37Tyr | Ins | |||||
| 1007-3338 | Fq Cap Cs Lzd Bdq | Arg255Pro | Ins + Del | |||||
| 1197-3582 | H E Z Pto | |||||||
| 1726-1727 | N/A | |||||||
| 1728-1729 | N/A | |||||||
| 2254-1349 | R E Z Fq Cap PAS Cs Lzd Trd Amc | Arg255Pro | ||||||
| 2297-4397 | N/A | |||||||
| 2371-4063 | R Fq Amg Cs Pto | Ser443Ala | ||||||
| 2619-5443 | N/A | Ins | Arg255Pro | Pro7Arg | ||||
| 2816-4888 | H R Fq Amg Lzd | |||||||
| 2961-3886 | Fq Lzd Pto Bdq | |||||||
| 3175-4904 | H R Z Fq | Gly119Ala | ||||||
| 3783-9 | Fq Amg PAS Cs Lzd Mrp | Ser443Ala | ||||||
| 3792-4254 | Z Fq Lzd Pto Bdq | Ins | ||||||
| 4190-4558 | H R E Z | Ser443Ala | ||||||
| 4192-4082 | Fq Cap Cs Lzd Pto Bdq | Ins | Cys18Tyr | |||||
| 4343-736 | Fq Amg Lzd Bdq Amc | Ins | Ins | Ser443Ala | Cys18Tyr | |||
| 4405-73 | E Z Fq Cs Lzd Bdq Mrp | Ins | Pro7Arg | |||||
| 4736-449 | H R E Z | |||||||
| 4767-5202210042 | Z Fq Cap Cs PAS Pto | Ins | Ile16Asn + Ins | Ins + Del | ||||
| Fq Cap Cs Pto Lzd Bdq | ||||||||
| Fq Cap Cs Pto Mrp Amc | ||||||||
| H R E Cap Pto | ||||||||
| 4936-7011 | Z Fq Cap Cs Lzd Bdq Mrp | |||||||
| 5978-1012200030 | Z Fq Cs Lzd Bdq Im Amc | |||||||
| 6633-8054 | Fq Amg Cap Cs Lzd Bdq Mrp Trd | Ins | ||||||
| 7135-1102210069 | Z Fq Cap Cs Lzd Bdq Mrp Amc | Ins | Ins | |||||
| 7261-7896 | R Z Fq Amg Cs | |||||||
| 1811200088-2501210046 | Fq Cs Lzd Bdq Mrp | Cys18Tyr | Ins + Del | |||||
| 2312200044-5203210083 | Fq Lzd Pto Bdq Mrp Amc | Cys37Tyr + Pro7Arg | ||||||
| 0033-3060 | N/A | Ins | Cys37Tyr | |||||
| 3060-0385 | Ins | Cys37Tyr | ||||||
| 2966-1617 | R E Z Fq Pto | |||||||
| 1617-1621 | ||||||||
| 4459-7077 | Fq Cap PAS Cs Pto | Ins | ||||||
| 7077-1233 | Ins | Ser443Ala | Pro7Arg | Ins | ||||
| 3264-6976 | Z Fq Cap PAS Cs | |||||||
| 6976-256 | Ins | |||||||
| 3411-4675 | Z Fq PAS Cs Pto | Arg255Pro | ||||||
| 4675-6782 | Fq Amg Cs Lzd Bdq Mrp Amc | Arg255Pro | ||||||
| 6782-7634 | Fq Amg Cs Lzd Bdq Mrp Amc | Ins | Arg255Pro | Ins | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagutkin, D.; Panova, A.; Vinokurov, A.; Gracheva, A.; Samoilova, A.; Vasilyeva, I. Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment. Microorganisms 2022, 10, 1440. https://doi.org/10.3390/microorganisms10071440
Lagutkin D, Panova A, Vinokurov A, Gracheva A, Samoilova A, Vasilyeva I. Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment. Microorganisms. 2022; 10(7):1440. https://doi.org/10.3390/microorganisms10071440
Chicago/Turabian StyleLagutkin, Denis, Anna Panova, Anatoly Vinokurov, Alexandra Gracheva, Anastasia Samoilova, and Irina Vasilyeva. 2022. "Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment" Microorganisms 10, no. 7: 1440. https://doi.org/10.3390/microorganisms10071440
APA StyleLagutkin, D., Panova, A., Vinokurov, A., Gracheva, A., Samoilova, A., & Vasilyeva, I. (2022). Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment. Microorganisms, 10(7), 1440. https://doi.org/10.3390/microorganisms10071440