
The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials & Methods
2.1. Study Population
2.2. Sample Collection
2.3. DNA Extraction and 16S rRNA Gene Sequence Processing
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Baseline Characteristics
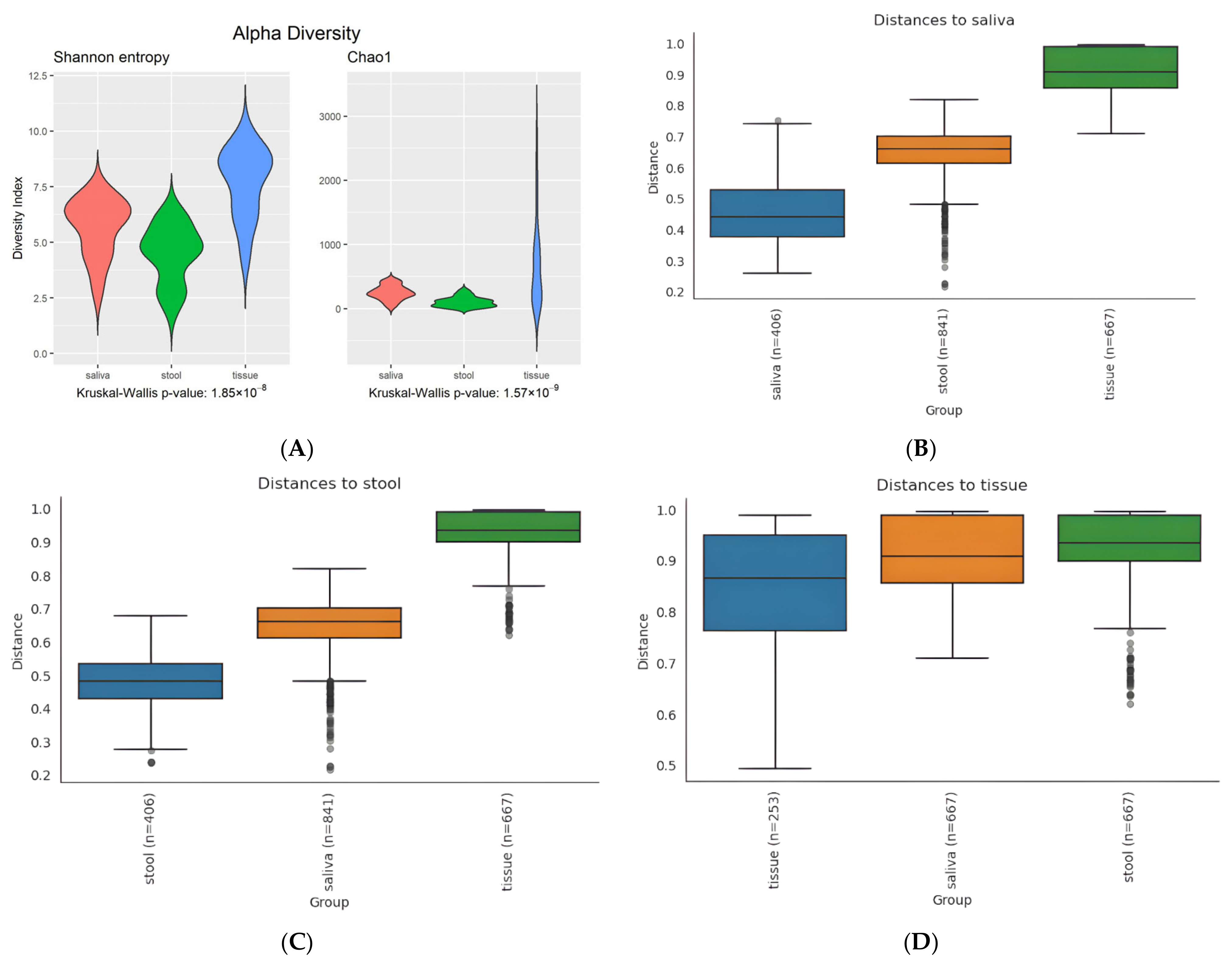
3.2. Diversity in Microbiota
3.2.1. Alpha Diversity
3.2.2. Beta Diversity
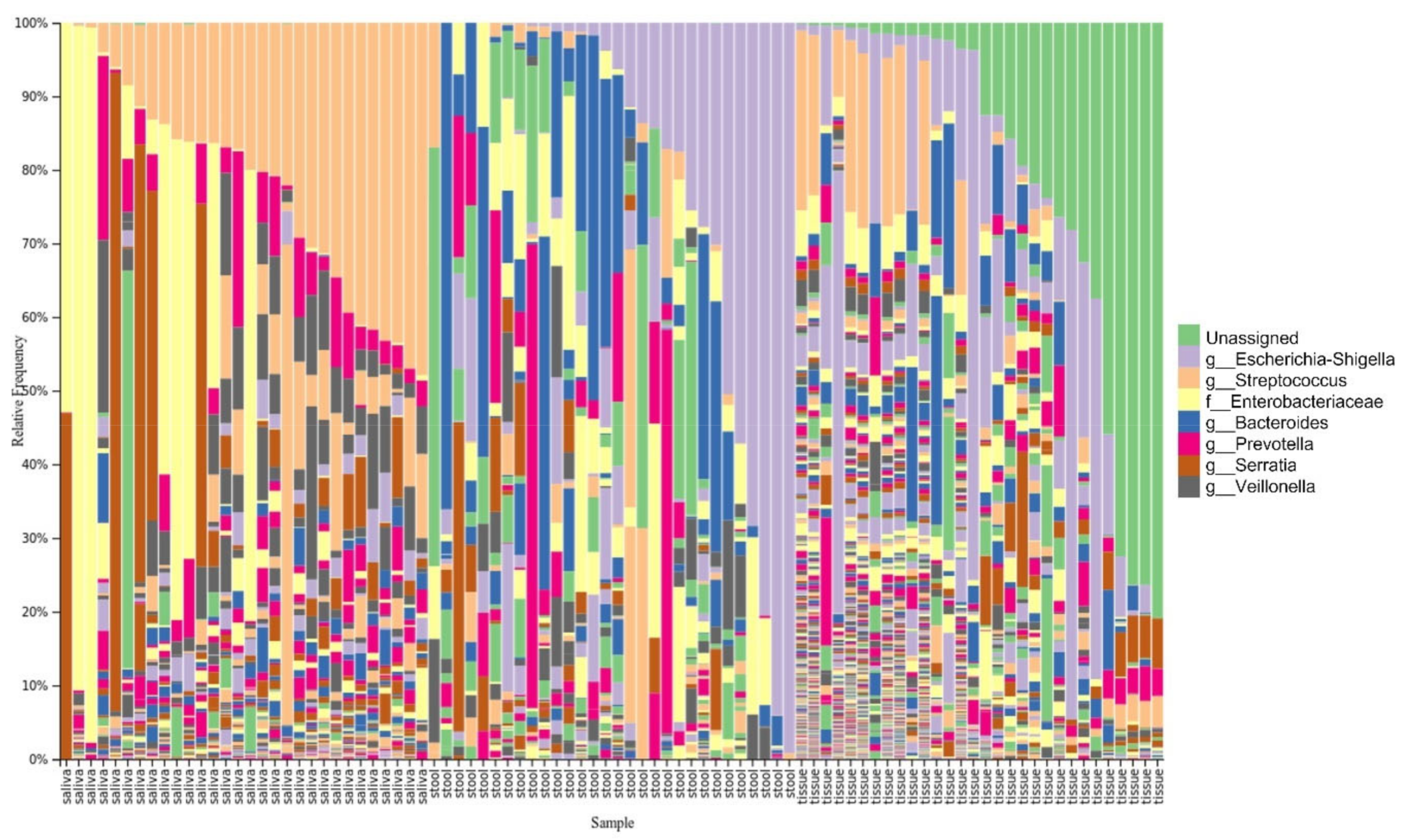
3.3. Taxonomy Distribution
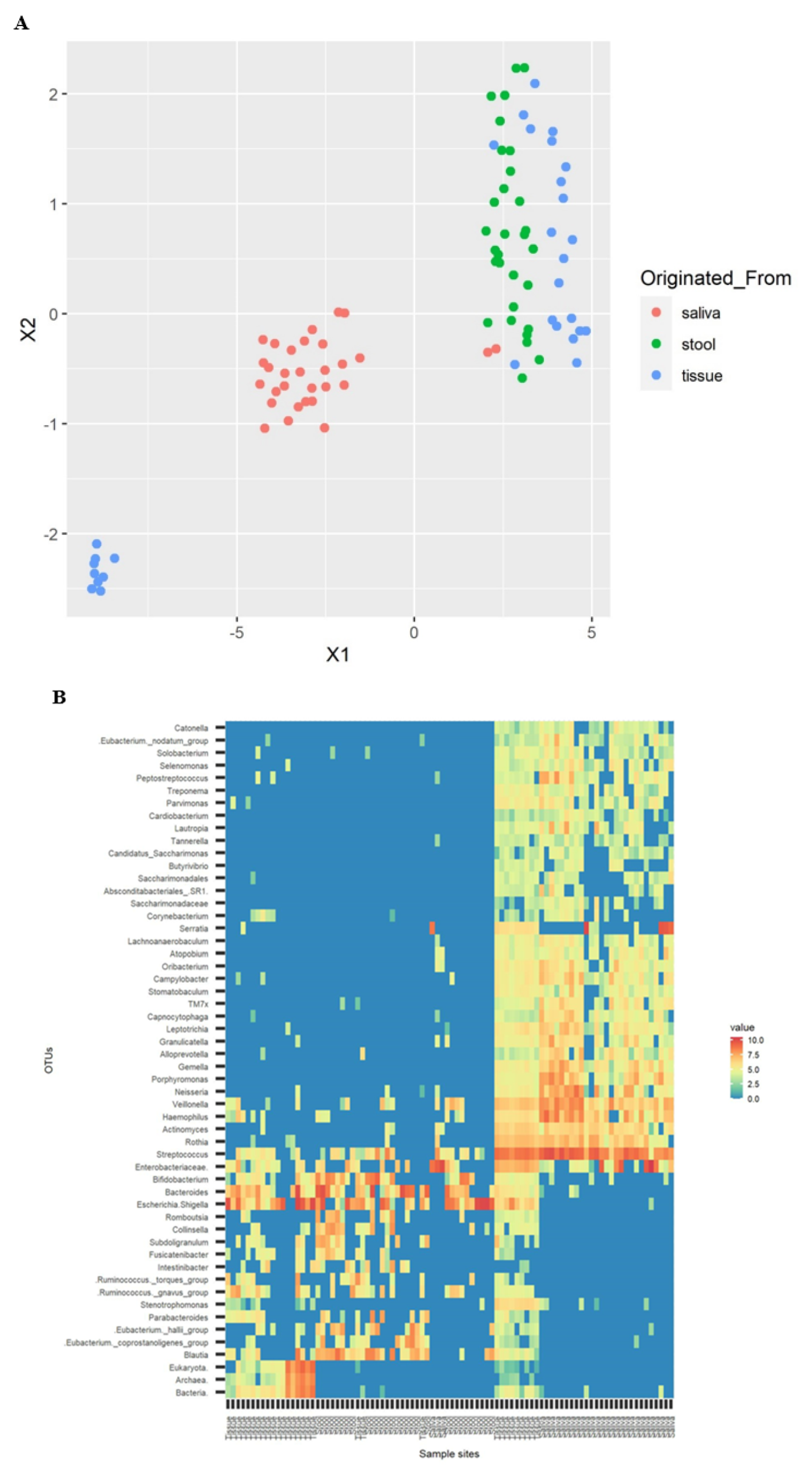
3.4. Cluster Visualization
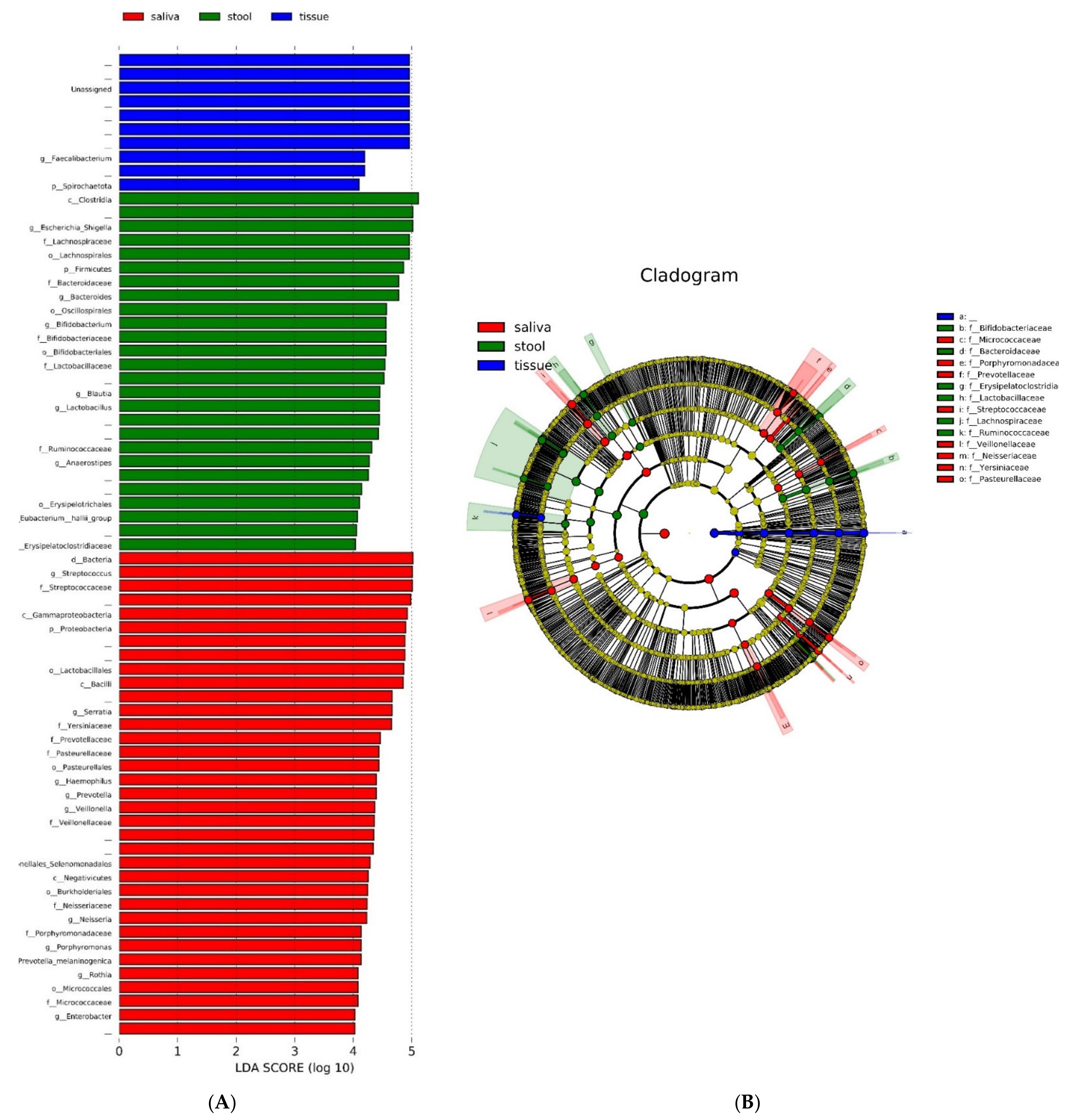
3.5. Quantitative and Phylogenic Analysis
3.6. Clinical Subgroup Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Low, D.; Ezaki, Y.; Okada, T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest Res. 2020, 18, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Lau, J.T.; Leontiadis, G.I.; Tse, F.; Yuan, Y.; Surette, M.; Moayyedi, P. Differences in gut microbiota in patients with vs. without inflammatory bowel diseases: A systematic review. Gastroenterology 2020, 158, 930–946.e931. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Brun, L.; Ballabeni, P.; Pittet, V.; Vavricka, B.M.P.; Zeitz, J.; Rogler, G.; Schoepfer, A.M.; The Swiss IBD Cohort Study Group. Frequency and risk factors for extraintestinal manifestations in the swiss inflammatory bowel disease cohort. Off. J. Am. Coll. Gastroenterol.|ACG 2011, 106, 110–119. [Google Scholar] [CrossRef]
- Baima, G.; Massano, A.; Squillace, E.; Caviglia, G.P.; Buduneli, N.; Ribaldone, D.G.; Aimetti, M. Shared microbiological and immunological patterns in periodontitis and ibd: A scoping review. Oral Dis. 2022, 28, 1029–1041. [Google Scholar] [CrossRef]
- Qi, Y.; Zang, S.-Q.; Wei, J.; Yu, H.-C.; Yang, Z.; Wu, H.-M.; Kang, Y.; Tao, H.; Yang, M.-F.; Jin, L.; et al. High-throughput sequencing provides insights into oral microbiota dysbiosis in association with inflammatory bowel disease. Genomics 2021, 113, 664–676. [Google Scholar] [CrossRef]
- Read, E.; Curtis, M.A.; Neves, J.F. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 731–742. [Google Scholar] [CrossRef]
- Plauth, M.; Jenss, H.; Meyle, J. Oral manifestations of crohn’s disease: An analysis of 79 cases. J. Clin. Gastroenterol. 1991, 13, 29–37. [Google Scholar] [CrossRef]
- Pittock, S.; Drumm, B.; Fleming, P.; McDermott, M.; Imrie, C.; Flint, S.; Bourke, B. The oral cavity in Crohn’s disease. J. Pediatr. 2001, 138, 767–771. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using qiime 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Sainburg, T.; McInnes, L.; Gentner, T.Q. Parametric umap embeddings for representation and semisupervised learning. Neural Comput. 2021, 33, 2881–2907. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, W.; Takeshita, T.; Shibata, Y.; Matsuo, K.; Eshima, N.; Yokoyama, T.; Yamashita, Y. Compositional stability of a salivary bacterial population against supragingival microbiota shift following periodontal therapy. PLoS ONE 2012, 7, e42806. [Google Scholar] [CrossRef] [Green Version]
- Paju, S.; Pussinen, P.J.; Suominen-Taipale, L.; Hyvönen, M.; Knuuttila, M.; Könönen, E. Detection of multiple pathogenic species in saliva is associated with periodontal infection in adults. J. Clin. Microbiol. 2009, 47, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.M. The immune response to prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Oakley, B.B.; Fiedler, T.L.; Marrazzo, J.M.; Fredricks, D.N. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl. Environ. Microbiol. 2008, 74, 4898–4909. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Gill, T.; Brooks, S.R.; Rosenbaum, J.T.; Asquith, M.; Colbert, R.A. Novel inter-omic analysis reveals relationships between diverse gut microbiota and host immune dysregulation in hla–b27–induced experimental spondyloarthritis. Arthritis Rheumatol. 2019, 71, 1849–1857. [Google Scholar] [CrossRef]
- Dinakaran, V.; Mandape, S.N.; Shuba, K.; Pratap, S.; Sakhare, S.S.; Tabatabai, M.A.; Smoot, D.T.; Farmer-Dixon, C.M.; Kesavalu, L.N.; Adunyah, S.E.; et al. Identification of specific oral and gut pathogens in full thickness colon of colitis patients: Implications for colon motility. Front. Microbiol. 2019, 9, 3220. [Google Scholar] [CrossRef] [PubMed]
- Said, H.S.; Suda, W.; Nakagome, S.; Chinen, H.; Oshima, K.; Kim, S.; Kimura, R.; Iraha, A.; Ishida, H.; Fujita, J.; et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2013, 21, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijakowski, K.; Surdacka, A. Salivary biomarkers for diagnosis of inflammatory bowel diseases: A systematic review. Int. J. Mol. Sci. 2020, 21, 7477. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.; Helmerhorst, E.J.; Gower, A.C.; Siqueira, W.L.; Paster, B.J.; Oppenheim, F.G.; Spormann, A.M. Microbial diversity in the early in vivo-formed dental biofilm. Appl. Environ. Microbiol. 2016, 82, 1881–1888. [Google Scholar] [CrossRef] [Green Version]
- Kaci, G.; Lakhdari, O.; Doré, J.; Ehrlich, S.D.; Renault, P.; Blottière, H.M.; Delorme, C. Inhibition of the NF-κB Pathway in Human Intestinal Epithelial Cells by Commensal Streptococcus salivarius. Appl. Environ. Microbiol. 2011, 77, 4681–4684. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Png, E.; Gowans, M.; Ong, D.E.H.; de Sessions, P.F.; Song, J.; Nagarajan, N. Ectopic gut colonization: A metagenomic study of the oral and gut microbiome in crohn’s disease. Gut Pathog. 2021, 13, 13. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Papa, E.; Docktor, M.; Smillie, C.; Weber, S.; Preheim, S.P.; Gevers, D.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Ingram, J.; et al. Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS ONE 2012, 7, e39242. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflamm. Bowel Dis. 2016, 22, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Jin, G.; Wang, G.; Liu, T.; Liu, X.; Wang, B.; Cao, H. Current sampling methods for gut microbiota: A call for more precise devices. Front. Cell. Infect. Microbiol. 2020, 10, 151. [Google Scholar] [CrossRef]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD (n = 30) | |
|---|---|
| Age (year) mean ± SD | 35.7 ± 11.2 |
| Male, n (%) | 21 (70.0) |
| BMI (kg/m2), mean ± SD | 22.4 ± 4.4 |
| Smoking status, n (%) | |
| Current | 4 (13.3) |
| Former | 1 (3.3) |
| Never | 25 (83.3) |
| Unknown | 0 (0.0) |
| Disease duration (year), mean ± SD | 6.8 ± 6.0 |
| Disease location, n (%) | |
| Ileum (L1) | 4 (13.3) |
| Colon (L2) | 8 (26.7) |
| Ileocolonic (L3) | 16 (53.3) |
| Ileocolonic (L3) + upper GI (L4) | 2 (6.7) |
| CDAI, mean ± SD | 52.7 ± 57.5 |
| Extraintestinal manifestation, n (%) | |
| Arthritis/arthralgia | 4 (13.3) |
| Uveitis/iritis | 0 (0.0) |
| Reactive skin lesion | 0 (0.0) |
| Concomitant drug use, n (%) | |
| 5-ASAs | 22 (73.3) |
| Corticosteroid | 16 (53.3) |
| Azathioprine/6-mercaptopurine | 0 (0.0) |
| Infliximab | 14 (46.7) |
| Adalimumab | 0 (0.0) |
| Ustekinumab | 2 (6.7) |
| Previous history of disease-related operations | 11 (36.7) |
| (A) | ||
|---|---|---|
| Genus | Rate of Containing Samples a | Total Relative Abundance b |
| Streptococcus | 96.67% | 22.29% |
| Serratia | 26.67% | 9.61% |
| Prevotella | 100.00% | 6.00% |
| Veillonella | 93.33% | 5.26% |
| Haemophilus | 83.33% | 4.83% |
| Neisseria | 83.33% | 3.43% |
| Porphyromonas | 86.67% | 2.63% |
| Rothia | 90.00% | 2.45% |
| Pantoea | 3.33% | 2.18% |
| Enterobacter | 16.67% | 2.07% |
| Actinomyces | 93.33% | 1.74% |
| Gemella | 90.00% | 1.68% |
| Fusobacterium | 83.33% | 1.58% |
| Campylobacter | 86.67% | 1.21% |
| Leptotrichia | 86.67% | 1.00% |
| Peptostreptococcus | 76.67% | 0.99% |
| Granulicatella | 80.00% | 0.96% |
| Alloprevotella | 80.00% | 0.91% |
| TM7x | 76.67% | 0.88% |
| Capnocytophaga | 86.67% | 0.77% |
| a Proportion of samples where one or more OTU count is detected. b Means of relative abundances of each OTU for samples. | ||
| (B) | ||
| Genus | Rate of Containing Samples | Total Relative Abundance |
| Escherichia Shigella | 93.33% | 13.47% |
| Streptococcus | 70.00% | 6.71% |
| Bacteroides | 90.00% | 3.91% |
| Faecalibacterium | 73.33% | 2.95% |
| Anaerostipes | 66.67% | 2.49% |
| Brachyspira | 3.33% | 2.00% |
| Ruminococcus gnavus group | 76.67% | 1.46% |
| Prevotella | 66.67% | 1.41% |
| Ruminococcus torques group | 46.67% | 1.14% |
| Lachnoclostridium | 63.33% | 1.08% |
| Rothia | 40.00% | 1.08% |
| Veillonella | 46.67% | 0.97% |
| Megamonas | 36.67% | 0.94% |
| Sutterella | 40.00% | 0.87% |
| Blautia | 70.00% | 0.77% |
| Bifidobacterium | 76.67% | 0.77% |
| Clostridium sensu stricto 1 | 56.67% | 0.75% |
| Fusobacterium | 63.33% | 0.72% |
| Actinomyces | 33.33% | 0.71% |
| Pseudomonas | 53.33% | 0.69% |
| (C) | ||
| Genus | Rate of Containing Samples | Total Relative Abundance |
| Escherichia Shigella | 80.00% | 20.80% |
| Bacteroides | 73.33% | 12.04% |
| Bifidobacterium | 63.33% | 7.04% |
| Lactobacillus | 33.33% | 5.69% |
| Blautia | 70.00% | 5.63% |
| Anaerostipes | 46.67% | 4.17% |
| Faecalibacterium | 46.67% | 2.29% |
| Eubacterium hallii group | 40.00% | 2.23% |
| Collinsella | 40.00% | 1.91% |
| Lachnoclostridium | 43.33% | 1.88% |
| Streptococcus | 56.67% | 1.79% |
| Eubacterium coprostanoligenes group | 40.00% | 1.78% |
| Prevotella | 26.67% | 1.73% |
| Megasphaera | 16.67% | 1.72% |
| Megamonas | 10.00% | 1.44% |
| Ruminococcus gnavus group | 36.67% | 1.31% |
| Romboutsia | 36.67% | 1.30% |
| Morganella | 6.67% | 1.20% |
| Pediococcus | 10.00% | 1.09% |
| Subdoligranulum | 30.00% | 1.08% |
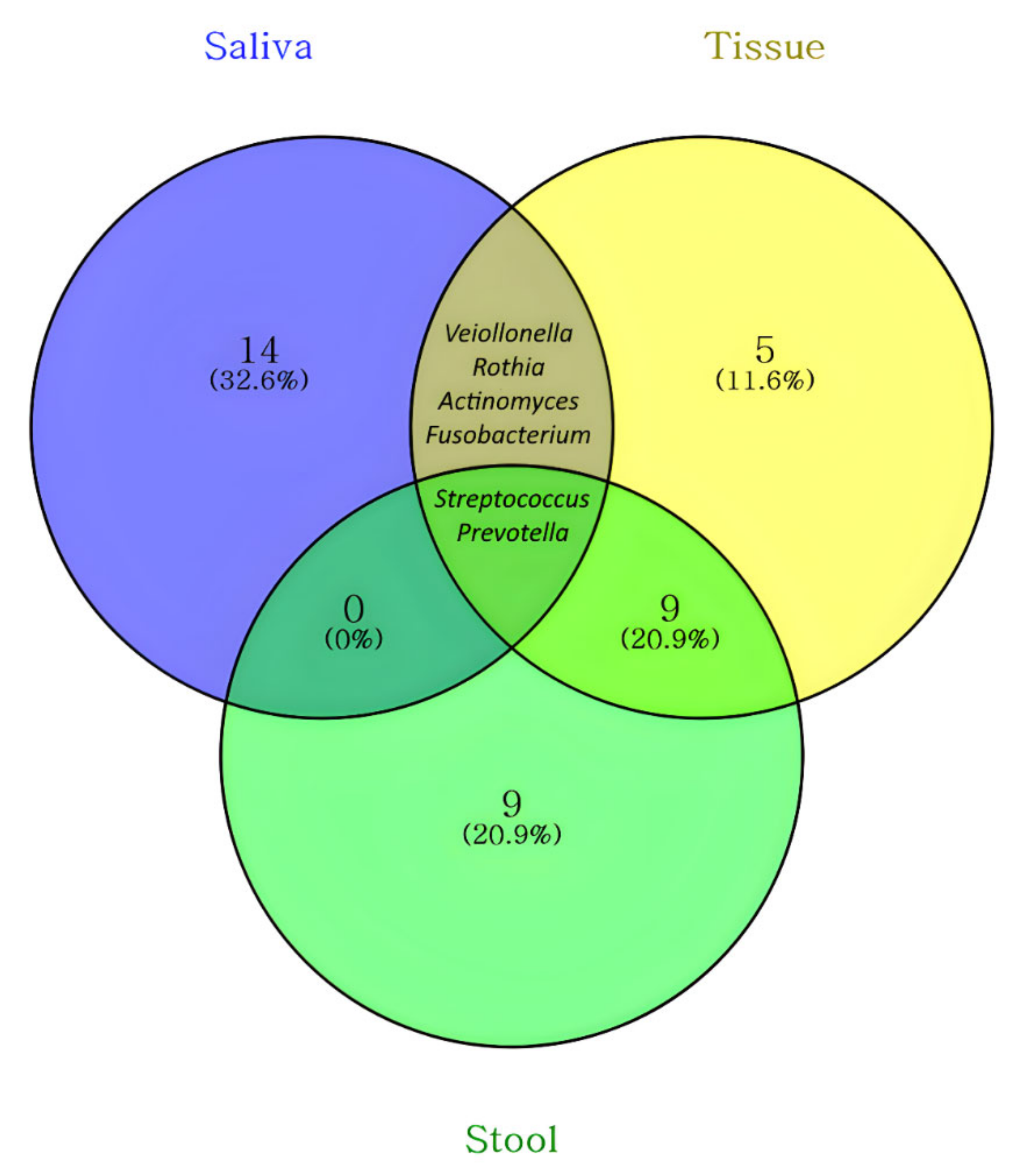
| Common Sites | Genus | Exclusive Sites | Genus |
|---|---|---|---|
| Saliva, tissue, and stool | Prevotella | Saliva | Alloprevotella |
| Streptococcus | Campylobacter | ||
| Saliva and tissue | Actinomyces | Capnocytophaga | |
| Fusobacterium | Enterobacter | ||
| Rothia | Gemella | ||
| Veillonella | Granulicatella | ||
| Tissue and stool | Ruminococcus gnavus group | Haemophilus | |
| Anaerostipes | Leptotrichia | ||
| Bacteroides | Neisseria | ||
| Bifidobacterium | Pantoea | ||
| Blautia | Peptostreptococcus | ||
| Escherichia Shigella | Porphyromonas | ||
| Faecalibacterium | Serratia | ||
| Lachnoclostridium | TM7x | ||
| Megamonas | Tissue | Ruminococcus torques group | |
| Brachyspira | |||
| Clostridium sensu stricto 1 | |||
| Pseudomonas | |||
| Sutterella | |||
| Stool | Eubacterium coprostanoligenes group | ||
| Eubacterium hallii group | |||
| Collinsella | |||
| Lactobacillus | |||
| Megasphaera | |||
| Morganella | |||
| Pediococcus | |||
| Romboutsia | |||
| Subdoligranulum |
| Site | Genus | Sex | Behavior (1,2,3) a | Behavior (Perianal) b | Age Group c | Location (2 vs. 1, 3) d | Location (1,2,3) e |
|---|---|---|---|---|---|---|---|
| Saliva | Streptococcus | 2.30 × 10−1 | 7.39 × 10−1 | 8.35 × 10−1 | 2.71 × 10−1 | 2.16 × 10−2 f | 5.84 × 10−2 |
| Veillonella | 9.10 × 10−1 | 5.44 × 10−1 | 3.29 × 10−1 | 1.99 × 10−2 f | 9.25 × 10−1 | 3.71 × 10−1 | |
| Pantoea | 1.27 × 10−1 | 5.65 × 10−1 | 2.85 × 10−1 | 4.14 × 10−1 | 5.46 × 10−1 | 3.88 × 10−2 f | |
| TM7x | 4.80 × 10−1 | 5.80 × 10−1 | 6.45 × 10−1 | 3.69 × 10−2 f | 1.00 × 10−0 | 8.80 × 10−1 | |
| Actinomyces | 6.19 × 10−1 | 7.81 × 10−1 | 8.52 × 10−1 | 3.10 × 10−1 | 1.79 × 10−2 f | 4.80 × 10−2 f | |
| Stool | Escherichia.Shigella | 3.18 × 10−1 | 1.48 × 10−2 f | 1.94 × 10−2 f | 1.81 × 10−2 f | 5.40 × 10−1 | 7.09 × 10−1 |
| Bacteroides | 3.61 × 10−1 | 4.18 × 10−2 | 7.53 × 10−1 | 2.77 × 10−1 | 9.25 × 10−1 | 1.92 × 10−1 | |
| Bifidobacterium | 7.63 × 10−1 | 2.89 × 10−2 | 6.70 × 10−1 | 8.22 × 10−1 | 1.36 × 10−1 | 2.27 × 10−1 | |
| Megamonas | 8.28 × 10−1 | 1.60 × 10−1 | 5.54 × 10−2 | 1.76 × 10−1 | 2.96 × 10−3 f | 1.21 × 10−2 f | |
| Eubacterium coprostanoligenes group | 8.69 × 10−2 | 9.53 × 10−1 | 1.52 × 10−1 | 2.38 × 10−2 f | 8.12 × 10−1 | 5.24 × 10−1 | |
| Megasphaera | 7.01 × 10−1 | 3.21 × 10−1 | 2.50 × 10−2 f | 6.84 × 10−1 | 3.48 × 10−1 | 5.32 × 10−1 | |
| Lachnoclostridium | 7.44 × 10−3 f | 1.69 × 10−1 | 2.32 × 10−1 | 2.63 × 10−1 | 3.51 × 10−1 | 3.04 × 10−1 | |
| Eubacterium hallii group | 3.19 × 10−1 | 5.48 × 10−1 | 2.15 × 10−2 f | 8.82 × 10−1 | 4.27 × 10−1 | 2.35 × 10−1 | |
| Collinsella | 7.59 × 10−1 | 5.04 × 10−1 | 3.73 × 10−1 | 4.41 × 10−1 | 2.62 × 10−2 f | 7.95 × 10−2 | |
| Tissue | Streptococcus | 1.04 × 10−3 f | 4.75 × 10−1 | 1.29 × 10−2 | 2.12 × 10−1 | 5.37 × 10−1 | 1.76 × 10−1 |
| Prevotella | 6.51 × 10−3 f | 8.21 × 10−1 | 5.67 × 10−1 | 2.62 × 10−1 | 2.14 × 10−1 | 4.55 × 10−1 | |
| Veillonella | 1.09 × 10−3 f | 8.71 × 10−1 | 2.41 × 10−1 | 6.85 × 10−1 | 1.40 × 10−1 | 1.96 × 10−1 | |
| Rothia | 4.64 × 10−4 f | 6.49 × 10−1 | 2.60 × 10−1 | 1.88 × 10−1 | 6.34 × 10−1 | 6.08 × 10−1 | |
| Faecalibacterium | 8.02 × 10−1 | 2.54 × 10−2 f | 1.93 × 10−1 | 5.20 × 10−1 | 6.83 × 10−2 | 1.45 × 10−1 | |
| Clostridium sensu stricto 1 | 3.57 × 10−2 f | 5.72 × 10−1 | 2.98 × 10−1 | 5.82 × 10−1 | 2.61 × 10−1 | 4.43 × 10−1 | |
| Megamonas | 1.35 × 10−1 | 2.29 × 10−1 | 4.85 × 10−2 f | 5.30 × 10−2 | 5.50 × 10−1 | 2.61 × 10−1 | |
| Actinomyces | 1.43 × 10−4 f | 9.52 × 10−1 | 2.76 × 10−1 | 2.60 × 10−1 | 1.89 × 10−1 | 1.06 × 10−1 | |
| Pseudomonas | 2.07 × 10−3 f | 7.02 × 10−1 | 2.26 × 10−2 f | 3.41 × 10−1 | 2.35 × 10−1 | 2.40 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.Y.; Kim, S.; Choi, J.W.; Kang, S.-B.; Kim, T.O.; Seo, G.S.; Cha, J.M.; Chun, J.; Jung, Y.; Im, J.P.; et al. The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease. Microorganisms 2022, 10, 1467. https://doi.org/10.3390/microorganisms10071467
Shin SY, Kim S, Choi JW, Kang S-B, Kim TO, Seo GS, Cha JM, Chun J, Jung Y, Im JP, et al. The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease. Microorganisms. 2022; 10(7):1467. https://doi.org/10.3390/microorganisms10071467
Chicago/Turabian StyleShin, Seung Yong, Sounkou Kim, Ji Won Choi, Sang-Bum Kang, Tae Oh Kim, Geom Seog Seo, Jae Myung Cha, Jaeyoung Chun, Yunho Jung, Jong Pil Im, and et al. 2022. "The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease" Microorganisms 10, no. 7: 1467. https://doi.org/10.3390/microorganisms10071467
APA StyleShin, S. Y., Kim, S., Choi, J. W., Kang, S. -B., Kim, T. O., Seo, G. S., Cha, J. M., Chun, J., Jung, Y., Im, J. P., Bang, K. B., Choi, C. H., Park, S. -K., & Park, D. I. (2022). The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease. Microorganisms, 10(7), 1467. https://doi.org/10.3390/microorganisms10071467