
Comparative Genomics and Physiology of Akkermansia muciniphila Isolates from Human Intestine Reveal Specialized Mucosal Adaptation

 , , , , ,
, , , , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Akkermansia muciniphila Isolates and Growth Conditions
2.2. Assessment of Sugar Fermentation Capacity
2.3. Proteome Analysis
2.4. Antibiotic Resistance Profiles
2.5. 16S rRNA Gene Sequence Identification
2.6. Phylogenetic Tree Construction
2.7. DNA Isolation and Genome Sequencing
2.8. Genome Annotation
2.9. Strain Identifiers and Accession Numbers
2.10. Statistical Analyses
3. Results
3.1. Enrichment and Isolation of Six Novel Akkermansia Strains from Human Feces
3.2. Growth Characteristics of the Six Akkermansia Isolates
3.3. Antibiotic Resistance Profiles of the Six A. muciniphila Isolates in Comparison with These of the Akkermansia Type-Strains
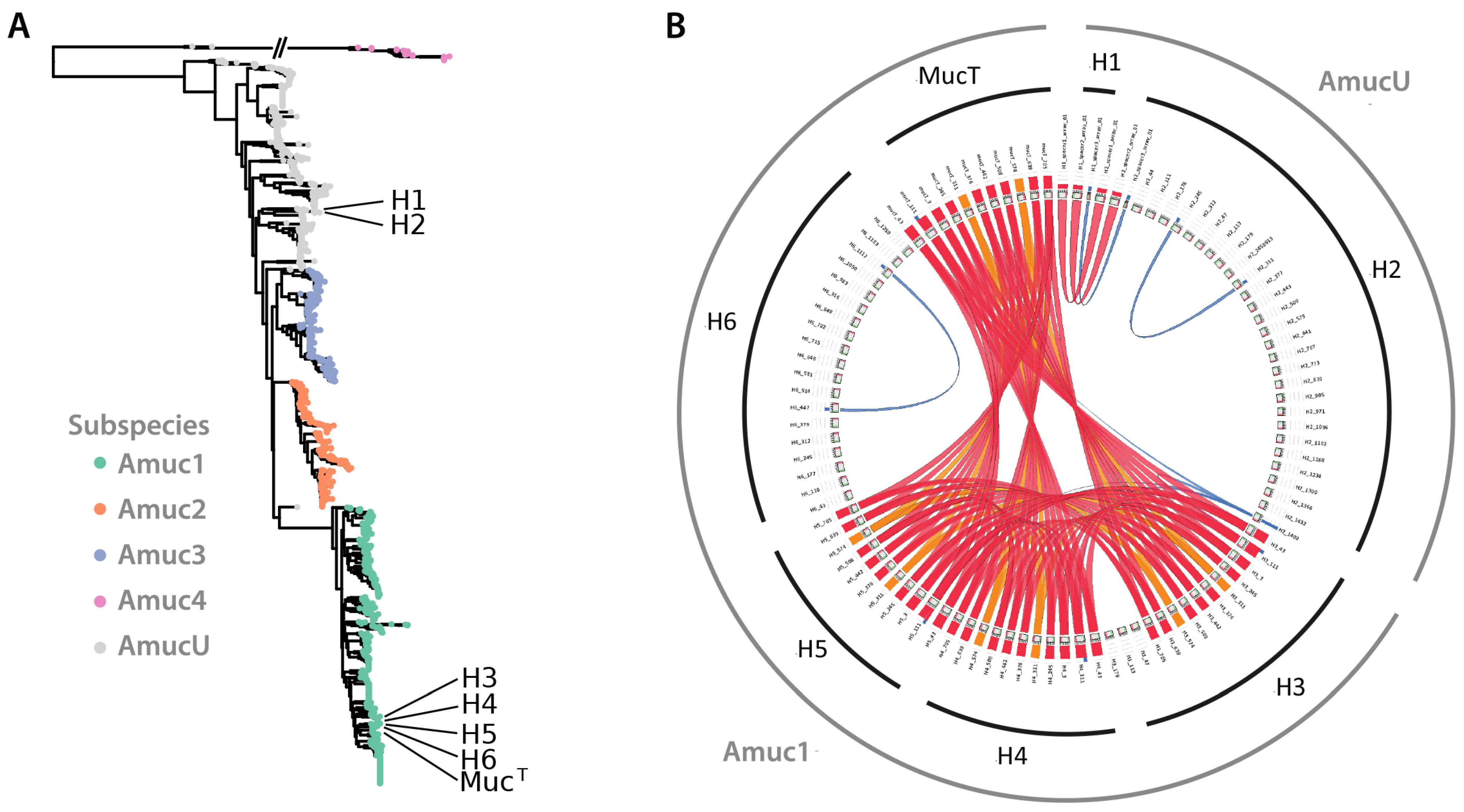
3.4. Comparative Genomics Confirms That the New Isolates Belong to Two Phylogenetic Clusters
3.5. Evaluation of CRISPR-Cas Systems in Novel A. muciniphila Isolates
3.6. Comparative Genomics Shows the Presence of Inversions and Rearrangements between Genomes of Strains H2 and MucT
3.7. Glycan-Degradation Capacity Is Conserved across All Isolates, but H2
3.8. Comparative Proteome Analysis of A. muciniphila Strain H2 and A. Glycaniphila
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.; Stecher, B.; Schintlmeister, A.; Reichert, J.; Brugiroux, S.; Wild, B.; Wanek, W.; Richter, A.; Rauch, I.; Decker, T.; et al. Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing. Proc. Natl. Acad. Sci. USA 2013, 110, 4720–4725. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Zoetendal, E.G.; von Wright, A.; Vilpponen-Salmela, T.; Ben-Amor, K.; Akkermans, A.D.; de Vos, W.M. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl. Environ. Microbiol. 2002, 68, 3401–3407. [Google Scholar] [CrossRef] [Green Version]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef]
- Belzer, C.; de Vos, W.M. Microbes inside—From diversity to function: The case of Akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Depommier, C.; Derrien, M.; Everard, A.; de Vos, W.M. Akkermansia muciniphila: Paradigm for next-generation beneficial microorganisms. Nat. Rev. Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Muller, M.; de Vos, W.M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [Green Version]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietila, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Van Hul, M. Novel opportunities for next-generation probiotics targeting metabolic syndrome. Curr. Opin. Biotechnol. 2015, 32, 21–27. [Google Scholar] [CrossRef]
- Depommier, C.; Van Hul, M.; Everard, A.; Delzenne, N.M.; De Vos, W.M.; Cani, P.D. Pasteurized Akkermansia muciniphila increases whole-body energy expenditure and fecal energy excretion in diet-induced obese mice. Gut Microbes 2020, 11, 1231–1245. [Google Scholar] [CrossRef] [Green Version]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- van Passel, M.W.; Kant, R.; Zoetendal, E.G.; Plugge, C.M.; Derrien, M.; Malfatti, S.A.; Chain, P.S.; Woyke, T.; Palva, A.; de Vos, W.M.; et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS ONE 2011, 6, e16876. [Google Scholar] [CrossRef] [Green Version]
- Ouwerkerk, J.P.; Aalvink, S.; Belzer, C.; de Vos, W.M. Akkermansia glycaniphila sp. nov., an anaerobic mucin-degrading bacterium isolated from reticulated python faeces. Int. J. Syst. Evol. Microbiol. 2016, 66, 4614–4620. [Google Scholar] [CrossRef]
- Ouwerkerk, J.P.; Koehorst, J.J.; Schaap, P.J.; Ritari, J.; Paulin, L.; Belzer, C.; de Vos, W.M. Complete Genome Sequence of Akkermansia glycaniphila Strain PytT, a Mucin-Degrading Specialist of the Reticulated Python Gut. Genome Announc. 2017, 5, e01098-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karcher, N.; Nigro, E.; Puncochar, M.; Blanco-Miguez, A.; Ciciani, M.; Manghi, P.; Zolfo, M.; Cumbo, F.; Manara, S.; Golzato, D.; et al. Genomic diversity and ecology of human-associated Akkermansia species in the gut microbiome revealed by extensive metagenomic assembly. Genome Biol. 2021, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; de Vos, W.M. Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front. Microbiol. 2017, 8, 1765. [Google Scholar] [CrossRef]
- EFSA Panel on Nutrition; Novel Foods and Food Allergens (NDA); Turck, D.; Bohn, T.; Castenmiller, J.; De Henauw, S.; Hirsch-Ernst, K.I.; Maciuk, A.; Mangelsdorf, I.; McArdle, H.J.; et al. Safety of pasteurised Akkermansia muciniphila as a novel food pursuant to Regulation (EU) 2015/2283. EFSA J. 2021, 18, e06135. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Belzer, C.; Goossens, M.; Kleerebezem, M.; De Vos, W.M.; Thas, O.; De Weirdt, R.; Kerckhof, F.M.; Van de Wiele, T. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013, 7, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.S.; Hoskins, L.C. Mucin degradation in human colon ecosystems. Fecal population densities of mucin-degrading bacteria estimated by a “most probable number” method. Gastroenterology 1981, 81, 759–765. [Google Scholar] [CrossRef]
- van Gelder, A.H.; Aydin, R.; Alves, M.M.; Stams, A.J. 1,3-Propanediol production from glycerol by a newly isolated Trichococcus strain. Microb. Biotechnol. 2012, 5, 573–578. [Google Scholar] [CrossRef] [Green Version]
- EFSA. Guidance on the assessment of bacterial susceptibility to antimicrobials of human and veterinary importance. EFSA J. 2012, 10, 2740. [Google Scholar] [CrossRef]
- Martin, R.; Jimenez, E.; Heilig, H.; Fernandez, L.; Marin, M.L.; Zoetendal, E.G.; Rodriguez, J.M. Isolation of bifidobacteria from breast milk and assessment of the bifidobacterial population by PCR-denaturing gradient gel electrophoresis and quantitative real-time PCR. Appl. Environ. Microbiol. 2009, 75, 965–969. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Peplies, J.; Glockner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salonen, A.; Nikkila, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilic-Stojanovic, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, K.; Paula, F.S.; Mueller, R.C.; Jesus Eda, C.; Cenciani, K.; Bohannan, B.J.; Nusslein, K.; Rodrigues, J.L. Forest-to-pasture conversion increases the diversity of the phylum Verrucomicrobia in Amazon rainforest soils. Front. Microbiol. 2015, 6, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, F.P.; Ribbera, A.; Kant, R.; Pietila, T.E.; Jarvinen, H.M.; Messing, M.; Randazzo, C.L.; Paulin, L.; Laine, P.; Ritari, J.; et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013, 9, e1003683. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, S.; Raymond, F.; Godzaridis, E.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt-Consortium. Activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claudel-Renard, C.; Chevalet, C.; Faraut, T.; Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003, 31, 6633–6639. [Google Scholar] [CrossRef] [Green Version]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, A.; Fineran, P.C.; Brown, C.M. Accurate computational prediction of the transcribed strand of CRISPR non-coding RNAs. Bioinformatics 2014, 30, 1805–1813. [Google Scholar] [CrossRef]
- Lange, S.J.; Alkhnbashi, O.S.; Rose, D.; Will, S.; Backofen, R. CRISPRmap: An automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 2013, 41, 8034–8044. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.C.; Derrien, M.; Isolauri, E.; de Vos, W.M.; Salminen, S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol. 2007, 73, 7767–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tindall, B.J.; Rossello-Mora, R.; Busse, H.J.; Ludwig, W.; Kampfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Bohme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [Green Version]
- Abbott, J.C.; Aanensen, D.M.; Bentley, S.D. WebACT: An online genome comparison suite. Methods Mol. Biol. 2007, 395, 57–74. [Google Scholar]
- Ottman, N.; Davids, M.; Suarez-Diez, M.; Boeren, S.; Schaap, P.J.; Martins Dos Santos, V.A.P.; Smidt, H.; Belzer, C.; de Vos, W.M. Genome-Scale Model and Omics Analysis of Metabolic Capacities of Akkermansia muciniphila Reveal a Preferential Mucin-Degrading Lifestyle. Appl. Environ. Microbiol. 2017, 83, e01014–e01017. [Google Scholar] [CrossRef] [Green Version]
- Kostopoulos, I.; Elzinga, J.; Ottman, N.; Klievink, J.T.; Blijenberg, B.; Aalvink, S.; Boeren, S.; Mank, M.; Knol, J.; de Vos, W.M.; et al. Akkermansia muciniphila uses human milk oligosaccharides to thrive in the early life conditions in vitro. Sci. Rep. 2020, 10, 14330. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Pollard, K.S. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallstrom, B.; Andersson, S.G. Genome reduction in the alpha-Proteobacteria. Curr. Opin. Microbiol. 2005, 8, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Kang, S.W.; Lee, J.H.; Park, S.H.; Lee, J.S. The evolution and competitive strategies of Akkermansia muciniphila in gut. Gut Microbes 2022, 14, 2025017. [Google Scholar] [CrossRef]
- Geerlings, S.Y.; Ouwerkerk, J.P.; Koehorst, J.J.; Ritari, J.; Aalvink, S.; Stecher, B.; Schaap, P.J.; Paulin, L.; de Vos, W.M.; Belzer, C. Genomic convergence between Akkermansia muciniphila in different mammalian hosts. BMC Microbiol. 2021, 21, 298. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Strain | Coverage | Contigs | Genome Size (Mbp) | GC Content (%) | Total Gene Count | ANI (%) | BLAST Similarity (>5 kb, %) | SNP |
|---|---|---|---|---|---|---|---|---|
| MucT | NA | 1 | 2.7 | 55.76 | 2178 | 100.00 | 100.00 | 0 |
| H1 | 60 | 34 | 2.8 | 55.04 | 2350 | 97.67 | 97.20 | 62,763 |
| H2 | 100 | 1 | 2.8 | 55.35 | 2348 | 97.62 | 97.16 | 62,508 |
| H3 | 120 | 30 | 2.7 | 56.05 | 2243 | 99.99 | 100.00 | 3 |
| H4 | 150 | 34 | 2.6 | 56.11 | 2204 | 99.99 | 100.00 | 3 |
| H5 | 150 | 18 | 2.5 | 55.80 | 2055 | 100.00 | 99.97 | 25 |
| H6 | 140 | 22 | 2.7 | 55.06 | 2208 | 99.99 | 99.88 | 27 |
| CAZy Family | GH 2 | GH 3 | GH 13 | GH 16 | GH 18 | GH 20 | GH 27 | GH 29 | GH 31 | GH 33 | GH 35 | GH 36 | GH 43 | GH 57 | GH 63 | GH 73 | GH 77 | GH 84 | GH 89 | GH 95 | GH 97 | GH 105 | GH 109 | GH 110 | GH 115 | GH 123 | GH 163 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MucT | 6 | 1 | 3 | 3 | 1 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| H1 | 5 | 1 | 3 | 3 | 0 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 1 | 1 | 1 | 0 | 1 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| H2 | 3 | 0 | 2 | 1 | 0 | 6 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| H3 | 6 | 1 | 3 | 3 | 1 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| H4 | 6 | 1 | 3 | 3 | 1 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| H5 | 6 | 1 | 3 | 3 | 1 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| H6 | 6 | 1 | 3 | 3 | 1 | 11 | 1 | 4 | 2 | 3 | 2 | 3 | 2 | 1 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 0 | 1 | 0 |
| PytT | 10 | 3 | 2 | 2 | 3 | 15 | 1 | 7 | 2 | 6 | 1 | 6 | 1 | 1 | 1 | 2 | 1 | 1 | 3 | 3 | 0 | 0 | 1 | 2 | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouwerkerk, J.P.; Tytgat, H.L.P.; Elzinga, J.; Koehorst, J.; Van den Abbeele, P.; Henrissat, B.; Gueimonde, M.; Cani, P.D.; Van de Wiele, T.; Belzer, C.; et al. Comparative Genomics and Physiology of Akkermansia muciniphila Isolates from Human Intestine Reveal Specialized Mucosal Adaptation. Microorganisms 2022, 10, 1605. https://doi.org/10.3390/microorganisms10081605
Ouwerkerk JP, Tytgat HLP, Elzinga J, Koehorst J, Van den Abbeele P, Henrissat B, Gueimonde M, Cani PD, Van de Wiele T, Belzer C, et al. Comparative Genomics and Physiology of Akkermansia muciniphila Isolates from Human Intestine Reveal Specialized Mucosal Adaptation. Microorganisms. 2022; 10(8):1605. https://doi.org/10.3390/microorganisms10081605
Chicago/Turabian StyleOuwerkerk, Janneke P., Hanne L. P. Tytgat, Janneke Elzinga, Jasper Koehorst, Pieter Van den Abbeele, Bernard Henrissat, Miguel Gueimonde, Patrice D. Cani, Tom Van de Wiele, Clara Belzer, and et al. 2022. "Comparative Genomics and Physiology of Akkermansia muciniphila Isolates from Human Intestine Reveal Specialized Mucosal Adaptation" Microorganisms 10, no. 8: 1605. https://doi.org/10.3390/microorganisms10081605
APA StyleOuwerkerk, J. P., Tytgat, H. L. P., Elzinga, J., Koehorst, J., Van den Abbeele, P., Henrissat, B., Gueimonde, M., Cani, P. D., Van de Wiele, T., Belzer, C., & de Vos, W. M. (2022). Comparative Genomics and Physiology of Akkermansia muciniphila Isolates from Human Intestine Reveal Specialized Mucosal Adaptation. Microorganisms, 10(8), 1605. https://doi.org/10.3390/microorganisms10081605