
Genomic Characterization of Salmonella Typhimurium Isolated from Guinea Pigs with Salmonellosis in Lima, Peru

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Bacterial Isolates
Whole Genome Sequencing and Assembly
2.2. Phylogenetic and Genetic Diversity Analysis of S. Typhimurium from Guinea Pigs
2.3. MLST, Serotype, Prophage, Plasmid, and Antibiotic Resistance Genes [ARGs] Profiles Prediction and Comparative Genomics
3. Results
3.1. Genomic Characteristics and Diversity of S. Typhimurium Isolates Obtained from Guinea Pigs
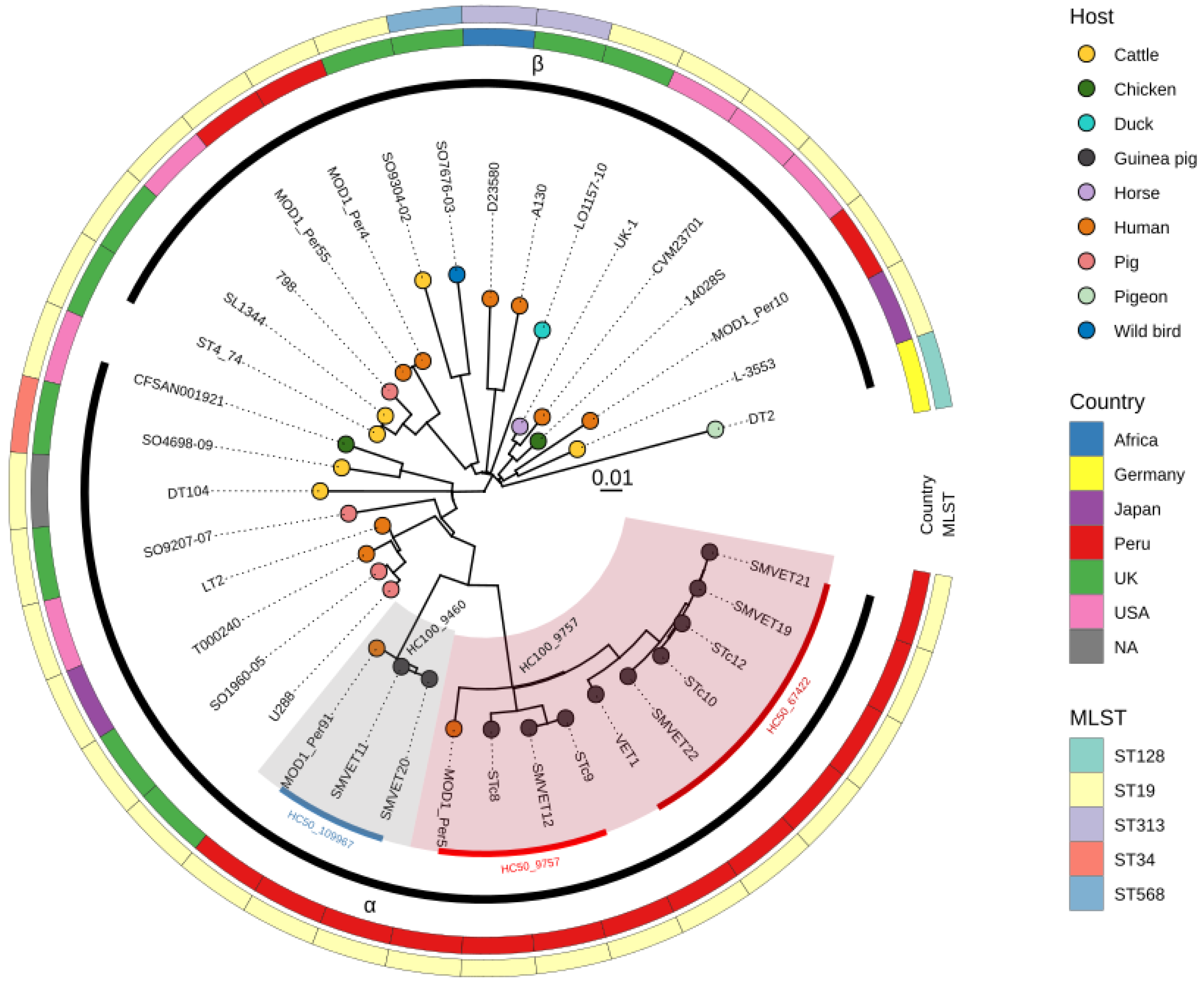
3.2. Phylogenomic Analysis of S. Typhimurium Isolated in Peru
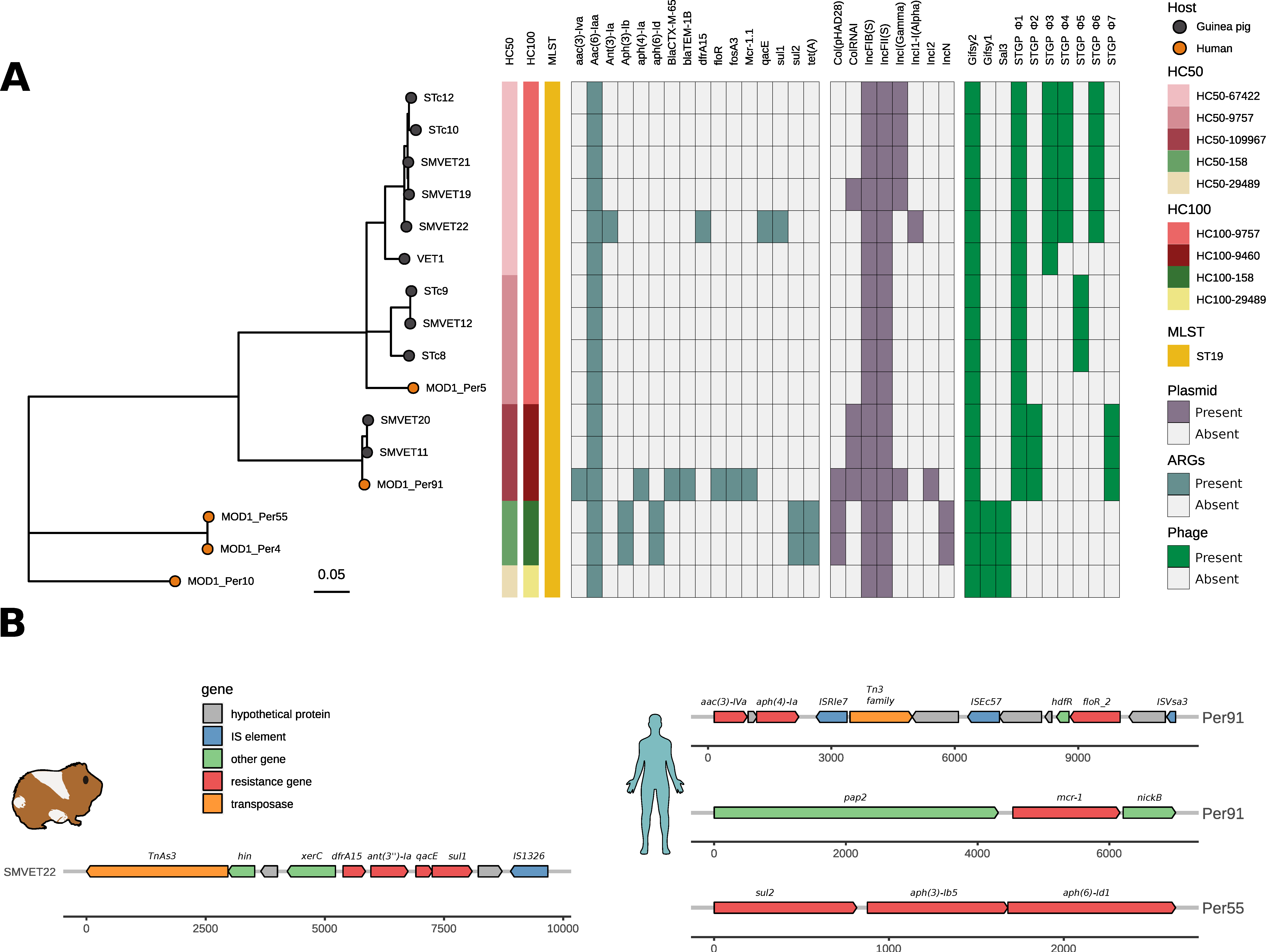
3.3. ARGs, Plasmid, and Virulence Factor Repertoire of S. Typhimurium from Guinea Pigs and Humans in Peru
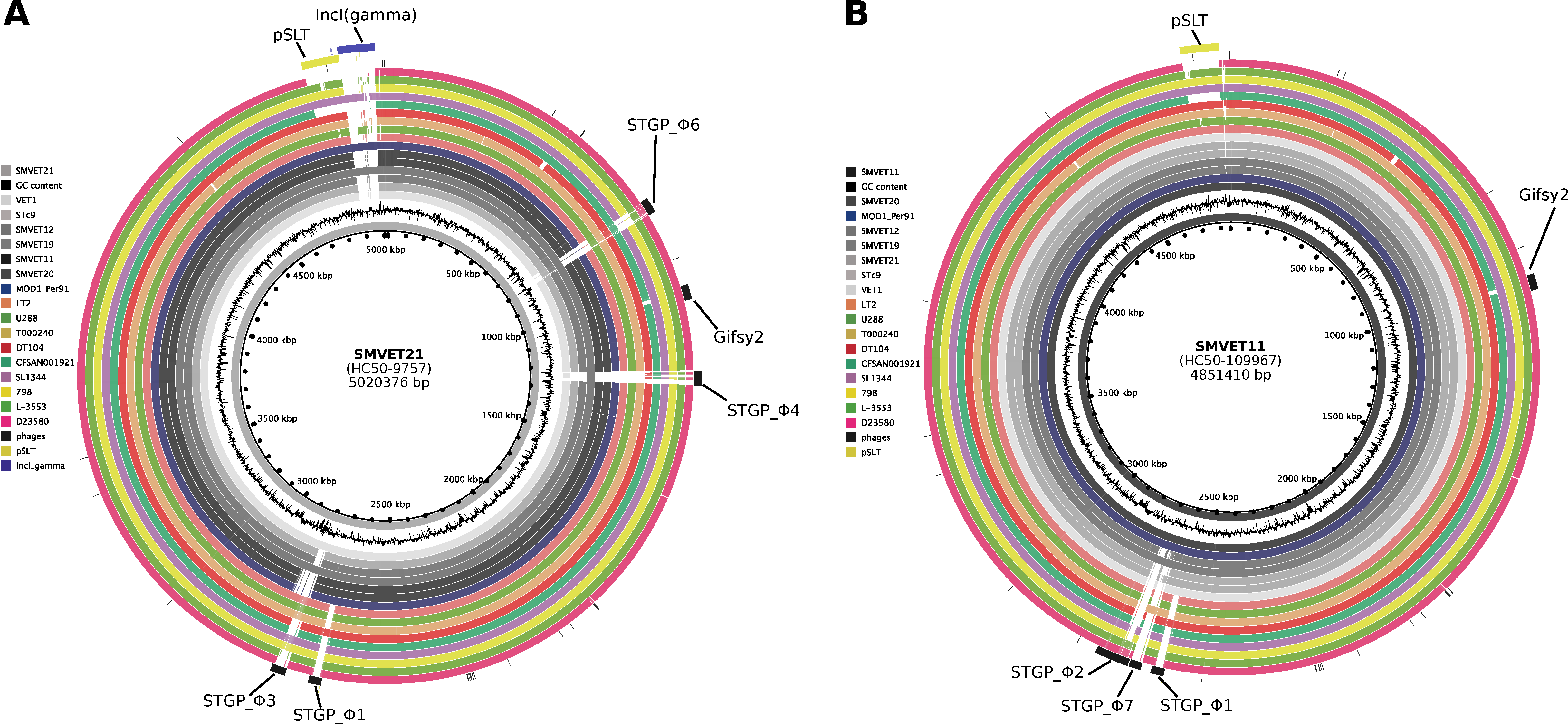
3.4. Differential Gene Content in S. Typhimurium Isolates from Guinea Pigs Is Driven by Phages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. The European Union Summary Report on Trends and Sources of Zoonoses, Zoonotic Agents and Food-Borne Outbreaks in 2016. EFSA J. 2017, 15, e05077. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention, National Salmonella Surveillance Annual Report. 2016. Available online: https://www.cdc.gov/nationalsurveillance/salmonella-surveillance.html (accessed on 3 August 2022).
- Branchu, P.; Bawn, M.; Kingsley, R.A. Genome Variation and Molecular Epidemiology of Salmonella Enterica Serovar Typhimurium Pathovariants. Infect. Immun. 2018, 86, e00079-18. [Google Scholar] [CrossRef] [PubMed]
- Okoro, C.K.; Barquist, L.; Connor, T.R.; Harris, S.R.; Clare, S.; Stevens, M.P.; Arends, M.J.; Hale, C.; Kane, L.; Pickard, D.J.; et al. Signatures of Adaptation in Human Invasive Salmonella Typhimurium ST313 Populations from Sub-Saharan Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003611. [Google Scholar] [CrossRef]
- Bawn, M.; Alikhan, N.-F.; Thilliez, G.; Kirkwood, M.; Wheeler, N.E.; Petrovska, L.; Dallman, T.J.; Adriaenssens, E.M.; Hall, N.; Kingsley, R.A. Evolution of Salmonella Enterica Serotype Typhimurium Driven by Anthropogenic Selection and Niche Adaptation. PLoS Genet. 2020, 16, e1008850. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, M.; Vohra, P.; Bawn, M.; Thilliez, G.; Pye, H.; Tanner, J.; Chintoan-Uta, C.; Branchu, P.; Petrovska, L.; Dallman, T.; et al. Ecological Niche Adaptation of Salmonella Typhimurium U288 Is Associated with Altered Pathogenicity and Reduced Zoonotic Potential. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Arrieta-Gisasola, A.; Atxaerandio-Landa, A.; Garrido, V.; Grilló, M.J.; Martínez-Ballesteros, I.; Laorden, L.; Garaizar, J.; Bikandi, J. Genotyping Study of Salmonella 4,[5],12:I:- Monophasic Variant of Serovar Typhimurium and Characterization of the Second-Phase Flagellar Deletion by Whole Genome Sequencing. Microorganisms 2020, 8, 2049. [Google Scholar] [CrossRef]
- Sun, H.; Wan, Y.; Du, P.; Bai, L. The Epidemiology of Monophasic Salmonella Typhimurium. Foodborne Pathog. Dis. 2020, 17, 87–97. [Google Scholar] [CrossRef]
- Gardy, J.L.; Loman, N.J. Towards a Genomics-Informed, Real-Time, Global Pathogen Surveillance System. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Argimón, S.; Yeats, C.A.; Goater, R.J.; Abudahab, K.; Taylor, B.; Underwood, A.; Sánchez-Busó, L.; Wong, V.K.; Dyson, Z.A.; Nair, S.; et al. A Global Resource for Genomic Predictions of Antimicrobial Resistance and Surveillance of Salmonella Typhi at Pathogenwatch. Nat. Commun. 2021, 12, 2879. [Google Scholar] [CrossRef]
- Lord, E.; Collins, C.; deFrance, S.; LeFebvre, M.J.; Pigière, F.; Eeckhout, P.; Erauw, C.; Fitzpatrick, S.M.; Healy, P.F.; Martínez-Polanco, M.F.; et al. Ancient DNA of Guinea Pigs (Cavia Spp.) Indicates a Probable New Center of Domestication and Pathways of Global Distribution. Sci. Rep. 2020, 10, 8901. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Macías, D.; Barba-Maggi, L.; Morales-de la Nuez, A.; Palmay-Paredes, J. Guinea Pig for Meat Production: A Systematic Review of Factors Affecting the Production, Carcass and Meat Quality. Meat Sci. 2018, 143, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; Guttman, D.S.; Fitzgerald, J.R. Population Genomics of Bacterial Host Adaptation. Nat. Rev. Genet. 2018, 19, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Mourkas, E.; Taylor, A.J.; Méric, G.; Bayliss, S.C.; Pascoe, B.; Mageiros, L.; Calland, J.K.; Hitchings, M.D.; Ridley, A.; Vidal, A.; et al. Agricultural Intensification and the Evolution of Host Specialism in the Enteric Pathogen Campylobacter Jejuni. Proc. Natl. Acad. Sci. USA 2020, 117, 11018–11028. [Google Scholar] [CrossRef]
- Matsuura, S.A.; Morales, C.S.; Calle, E.S.; Ara, G.M. Susceptibilidad a Antibacterianos in Vitro de Salmonella Enterica Aislada de Cuyes de Crianza Familiar-Comercial En La Provincia de Carhuaz, Áncash. Rev. Investig. Vet. Perú 2010, 21, 93–99. [Google Scholar] [CrossRef]
- Moya, A.L. Prevalencia de Salmonelosis en Cuyes (Cavia porcellus) Procedentes de Granjas del Centro Poblado Huancaquito Alto–Virú–La Libertad; Universidad Privada Antenor Orrego–UPAO: Trujillo, Peru, 2019. [Google Scholar]
- Fournier, J.B.; Knox, K.; Harris, M.; Newstein, M. Family Outbreaks of Nontyphoidal Salmonellosis Following a Meal of Guinea Pigs. Case Rep. Infect. Dis. 2015, 2015, e864640. [Google Scholar] [CrossRef]
- Marcelo, M.G.; Rosadio, A.R.; Chero, O.A.; Díaz, O.G.; Ciprian, C.A.; Maturrano, H.L. Identificación de Salmonella Enteritidis y Salmonella Typhimurium En Cuyes Mediante La Técnica de PCR Múltiple. Rev. Investig. Vet. Perú 2017, 28, 411–417. [Google Scholar] [CrossRef]
- Ikhimiukor, O.O.; Odih, E.E.; Donado-Godoy, P.; Okeke, I.N. A Bottom-up View of Antimicrobial Resistance Transmission in Developing Countries. Nat. Microbiol. 2022, 7, 757–765. [Google Scholar] [CrossRef]
- Jamshidi, A.; Kalidari, G.A.; Hedayati, M. Isolation and Identification of Salmonella Enteritidis and Salmonella Typhimurium from the Eggs of Retail Stores in Mashhad, Iran Using Conventional Culture Method and Multiplex Pcr Assay. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1745-4565.2010.00225.x (accessed on 6 August 2022).
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 May 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid Phylogenetic Analysis of Large Samples of Recombinant Bacterial Whole Genome Sequences Using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Zhang, S.; den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef]
- Banerji, S.; Simon, S.; Tille, A.; Fruth, A.; Flieger, A. Genome-Based Salmonella Serotyping as the New Gold Standard. Sci. Rep. 2020, 10, 4333. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 1st ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Chauca, L. Producción de Cuyes (Cavia Porcellus); FAO: Rome, Italy, 1997. [Google Scholar]
- Salvatierra, R.G.; Rimac, B.R.; Chero, O.A.; Reyna, W.I.; Rosadio, A.R.; Maturrano, H.L. Resistencia Antimicrobiana y Genotipificación de Cepas de Salmonella Typhimurium Aisladas de Cuyes (Cavia Porcellus) Provenientes de Granjas de Producción Intensiva de La Ciudad de Lima, Perú. Rev. Investig. Vet. Perú 2018, 29, 319–327. [Google Scholar] [CrossRef]
- Duran Gonzalez, C.; Luna Espinoza, L.; Carhuaricra Huamán, D.; Salvatierra Rodríguez, G.; Rosadio Alcántara, R.; Maturrano Hernández, L.; Duran Gonzalez, C.; Luna Espinoza, L.; Carhuaricra Huamán, D.; Salvatierra Rodríguez, G.; et al. Evaluación de Factores de Virulencia En Cepas de Salmonella Typhimurium Aisladas de Cuyes (Cavia Porcellus) Enfermos y Sanos. Rev. Investig. Vet. Perú 2021, 32, e21331. [Google Scholar] [CrossRef]
- Ido, N.; Lee, K.; Iwabuchi, K.; Izumiya, H.; Uchida, I.; Kusumoto, M.; Iwata, T.; Ohnishi, M.; Akiba, M. Characteristics of Salmonella Enterica Serovar 4,[5],12:I:- As a Monophasic Variant of Serovar Typhimurium. PLoS ONE 2014, 9, e104380. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Arias, P.; Montaño, L.A.; Villarreal, J.M.; Wiesner, M. Molecular and Phenotypic Characterization of Salmonella Typhimurium Monophasic Variant (1,4,[5],12:I:-) From Colombian Clinical Isolates. Biomed. Rev. Inst. Nac. Salud 2020, 40, 722–733. [Google Scholar] [CrossRef]
- Huamán, M.; Pérez, C.; Rodríguez, J.; Killerby, M.; Lovón, S.; Chauca, L.; Huamán, M.; Pérez, C.; Rodríguez, J.; Killerby, M.; et al. Caracterización Genética y Patrones de Resistencia Antimicrobiana En Cepas de Salmonella Enterica Subsp. Enterica Serovar Typhimurium En Cuyes de Crianza Intensiva. Rev. Investig. Vet. Perú 2020, 31, e17542. [Google Scholar] [CrossRef]
- Robertson, S.; Burakoff, A.; Stevenson, L.; Tompkins, B.; Patel, K.; Tolar, B.; Whitlock, L.; House, J.; Schlater, L.; Mackie, T.; et al. Notes from the Field: Recurrence of a Multistate Outbreak of Salmonella Enteritidis Infections Linked to Contact with Guinea Pigs–Eight States, 2015–2017. Morb. Mortal. Wkly. Rep. 2018, 67, 1195–1196. [Google Scholar] [CrossRef] [PubMed]
- Dróżdż, M.; Małaszczuk, M.; Paluch, E.; Pawlak, A. Zoonotic Potential and Prevalence of Salmonella Serovars Isolated from Pets. Infect. Ecol. Epidemiol. 2021, 11, 1975530. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, S.W.M.; Orsel, K.; Wagenaar, J.A.; Miko, A.; van Duijkeren, E. Animal-to-Human Transmission of Salmonella Typhimurium DT104A Variant. Emerg. Infect. Dis. 2004, 10, 2225–2227. [Google Scholar] [CrossRef]
- Stevens, M.P.; Humphrey, T.J.; Maskell, D.J. Molecular Insights into Farm Animal and Zoonotic Salmonella Infections. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2709–2723. [Google Scholar] [CrossRef]
- Lanzas, C.; Warnick, L.D.; James, K.L.; Wright, E.M.; Wiedmann, M.; Gröhn, Y.T. Transmission Dynamics of a Multidrug-Resistant Salmonella Typhimurium Outbreak in a Dairy Farm. Foodborne Pathog. Dis. 2010, 7, 467–474. [Google Scholar] [CrossRef]
- Salipante, S.J.; Hall, B.G. Determining the Limits of the Evolutionary Potential of an Antibiotic Resistance Gene. Mol. Biol. Evol. 2003, 20, 653–659. [Google Scholar] [CrossRef]
- Leon, I.M.; Lawhon, S.D.; Norman, K.N.; Threadgill, D.S.; Ohta, N.; Vinasco, J.; Scott, H.M. Serotype Diversity and Antimicrobial Resistance among Salmonella Enterica Isolates from Patients at an Equine Referral Hospital. Appl. Environ. Microbiol. 2018, 84, e02829.17. [Google Scholar] [CrossRef] [Green Version]
- Almeida, F.; Seribelli, A.A.; Medeiros, M.I.C.; Rodrigues, D.D.P.; de MelloVarani, A.; Luo, Y.; Allard, M.W.; Falcão, J.P. Phylogenetic and Antimicrobial Resistance Gene Analysis of Salmonella Typhimurium Strains Isolated in Brazil by Whole Genome Sequencing. PLoS ONE 2018, 13, e0201882. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.L.; Panzenhagen, P.; Ferrari, R.G.; Dos Santos, A.; Paschoalin, V.M.F.; Conte-Junior, C.A. Frequency of Antimicrobial Resistance Genes in Salmonella from Brazil by in Silico Whole-Genome Sequencing Analysis: An Overview of the Last Four Decades. Front. Microbiol. 2020, 11, 1864. [Google Scholar] [CrossRef] [PubMed]
- Madge, D.S. Effect of Antibiotics on Intestinal Absorption in Guinea-Pigs. Comp. Biochem. Physiol. 1969, 30, 295–307. [Google Scholar] [CrossRef]
- Carroll, L.M.; Wiedmann, M.; den Bakker, H.; Siler, J.; Warchocki, S.; Kent, D.; Lyalina, S.; Davis, M.; Sischo, W.; Besser, T.; et al. Whole-Genome Sequencing of Drug-Resistant Salmonella Enterica Isolates from Dairy Cattle and Humans in New York and Washington States Reveals Source and Geographic Associations. Appl. Environ. Microbiol. 2017, 83, e00140-17. [Google Scholar] [CrossRef]
- Aljahdali, N.H.; Foley, S.L.; Han, J.; Sanad, Y.M.; Nayak, R.; Khajanchi, B.K. Whole-Genome Sequences of 66 Incompatibility Group FIB Plasmid-Carrying Salmonella Enterica Serovar Typhimurium Isolates from Food Animal Sources. Microbiol. Resour. Announc. 2020, 9, e01435-19. [Google Scholar] [CrossRef]
- Deng, Y.; He, L.; Chen, S.; Zheng, H.; Zeng, Z.; Liu, Y.; Sun, Y.; Ma, J.; Chen, Z.; Liu, J.-H. F33:A−:B− and F2:A−:B− Plasmids Mediate Dissemination of RmtB-BlaCTX-M-9 Group Genes and RmtB-QepA in Enterobacteriaceae Isolates from Pets in China. Antimicrob. Agents Chemother. 2011, 55, 4926–4929. [Google Scholar] [CrossRef]
- Han, J.; Lynne, A.M.; David, D.E.; Tang, H.; Xu, J.; Nayak, R.; Kaldhone, P.; Logue, C.M.; Foley, S.L. DNA Sequence Analysis of Plasmids from Multidrug Resistant Salmonella Enterica Serotype Heidelberg Isolates. PLoS ONE 2012, 7, e51160. [Google Scholar] [CrossRef]
- Carattoli, A.; Villa, L.; Fortini, D.; García-Fernández, A. Contemporary IncI1 Plasmids Involved in the Transmission and Spread of Antimicrobial Resistance in Enterobacteriaceae. Plasmid 2021, 118, 102392. [Google Scholar] [CrossRef]
- Miller, E.A.; Elnekave, E.; Flores-Figueroa, C.; Johnson, A.; Kearney, A.; Munoz-Aguayo, J.; Tagg, K.A.; Tschetter, L.; Weber, B.P.; Nadon, C.A.; et al. Emergence of a Novel Salmonella Enterica Serotype Reading Clonal Group Is Linked to Its Expansion in Commercial Turkey Production, Resulting in Unanticipated Human Illness in North America. mSphere 2020, 5, e00056-20. [Google Scholar] [CrossRef]
- Leverstein-van Hall, M.A.; Dierikx, C.M.; Cohen Stuart, J.; Voets, G.M.; van den Munckhof, M.P.; van Essen-Zandbergen, A.; Platteel, T.; Fluit, A.C.; van de Sande-Bruinsma, N.; Scharinga, J.; et al. Dutch Patients, Retail Chicken Meat and Poultry Share the Same ESBL Genes, Plasmids and Strains. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2011, 17, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.J.; Shepard, S.M.; Rivet, B.; Danzeisen, J.L.; Carattoli, A. Comparative Genomics and Phylogeny of the IncI1 Plasmids: A Common Plasmid Type among Porcine Enterotoxigenic Escherichia Coli. Plasmid 2011, 66, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Razavi, M.; Kristiansson, E.; Flach, C.-F.; Larsson, D.G.J. The Association between Insertion Sequences and Antibiotic Resistance Genes. mSphere 2020, 5, e00418-20. [Google Scholar] [CrossRef] [PubMed]
- Mottawea, W.; Duceppe, M.-O.; Dupras, A.A.; Usongo, V.; Jeukens, J.; Freschi, L.; Emond-Rheault, J.-G.; Hamel, J.; Kukavica-Ibrulj, I.; Boyle, B.; et al. Salmonella Enterica Prophage Sequence Profiles Reflect Genome Diversity and Can Be Used for High Discrimination Subtyping. Front. Microbiol. 2018, 9, 836. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.T.; Huynh, S.; Alexander, A.; Oliver, A.S.; Cooper, K.K. Genomic Characterization of Salmonella Typhimurium DT104 Strains Associated with Cattle and Beef Products. Pathogens 2021, 10, 529. [Google Scholar] [CrossRef]
- Gal-Mor, O.; Boyle, E.C.; Grassl, G.A. Same Species, Different Diseases: How and Why Typhoidal and Non-Typhoidal Salmonella Enterica Serovars Differ. Front. Microbiol. 2014, 5, 391. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Isolate | Biosample ID | Average Coverage | Number of Contigs | Number of Bases | Number of CDS | %GC | MLST | Completeness * | Contamination * | HierCC HC100 |
|---|---|---|---|---|---|---|---|---|---|---|
| SMVET11 | SAMN28944802 | 61.012 | 44 | 4,851,410 | 4514 | 52.23 | 19 | 100 | 0.39 | 9460 |
| SMVET12 | SAMN28944803 | 76.632 | 45 | 4,902,895 | 4571 | 52.14 | 19 | 100 | 0.08 | 9757 |
| SMVET19 | SAMN28944804 | 93.268 | 61 | 5,088,465 | 4780 | 52.08 | 19 | 100 | 0.39 | 9757 |
| SMVET20 | SAMN28944805 | 106.307 | 39 | 4,849,526 | 4514 | 52.23 | 19 | 100 | 0.39 | 9460 |
| SMVET21 | SAMN28944807 | 83.118 | 44 | 5,020,376 | 4699 | 52.10 | 19 | 100 | 0.08 | 9757 |
| SMVET22 | SAMN28944808 | 115.677 | 54 | 5,095,938 | 4803 | 52.07 | 19 | 100 | 0.08 | 9757 |
| STc10 | SAMN28944806 | 30.112 | 175 | 5,046,798 | 4725 | 52.22 | 19 | 100 | 0.08 | 9757 |
| STc12 | SAMN28944811 | 36.224 | 65 | 5,017,046 | 4694 | 52.13 | 19 | 100 | 0.08 | 9757 |
| STc8 | SAMN28944810 | 31.676 | 80 | 4,915,140 | 4563 | 52.09 | 19 | 99.69 | 0.15 | 9757 |
| STc9 | SAMN28944809 | 61.838 | 39 | 4,891,950 | 4548 | 52.15 | 19 | 100 | 0.08 | 9757 |
| VET1 | SAMN28944812 | 316.099 | 86 | 4,910,420 | 4557 | 52.12 | 19 | 100 | 0.9 | 9757 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carhuaricra Huaman, D.E.; Luna Espinoza, L.R.; Rodríguez Cueva, C.L.; Duran Gonzales, C.G.; Rosadio Alcántara, R.H.; Setubal, J.C.; Maturrano Hernández, L. Genomic Characterization of Salmonella Typhimurium Isolated from Guinea Pigs with Salmonellosis in Lima, Peru. Microorganisms 2022, 10, 1726. https://doi.org/10.3390/microorganisms10091726
Carhuaricra Huaman DE, Luna Espinoza LR, Rodríguez Cueva CL, Duran Gonzales CG, Rosadio Alcántara RH, Setubal JC, Maturrano Hernández L. Genomic Characterization of Salmonella Typhimurium Isolated from Guinea Pigs with Salmonellosis in Lima, Peru. Microorganisms. 2022; 10(9):1726. https://doi.org/10.3390/microorganisms10091726
Chicago/Turabian StyleCarhuaricra Huaman, Dennis E., Luis R. Luna Espinoza, Carmen L. Rodríguez Cueva, Carla G. Duran Gonzales, Raúl H. Rosadio Alcántara, João C. Setubal, and Lenin Maturrano Hernández. 2022. "Genomic Characterization of Salmonella Typhimurium Isolated from Guinea Pigs with Salmonellosis in Lima, Peru" Microorganisms 10, no. 9: 1726. https://doi.org/10.3390/microorganisms10091726
APA StyleCarhuaricra Huaman, D. E., Luna Espinoza, L. R., Rodríguez Cueva, C. L., Duran Gonzales, C. G., Rosadio Alcántara, R. H., Setubal, J. C., & Maturrano Hernández, L. (2022). Genomic Characterization of Salmonella Typhimurium Isolated from Guinea Pigs with Salmonellosis in Lima, Peru. Microorganisms, 10(9), 1726. https://doi.org/10.3390/microorganisms10091726