
Evaluation of Bacterial Composition and Viability of Equine Feces after Processing for Transplantation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.1.1. Donor Horse
2.1.2. Preparations before Carrying out the Procedure
2.1.3. Sample Processing, Collection and Storage
2.2. Propidium Monoazide (PMA) Treatment
2.3. Bacterial DNA Extraction and 16S Amplicon Sequencing
2.4. Sequence Analysis and 16S rDNA Profiling
2.5. Quantitative PCR
2.6. Data Analysis
3. Results
3.1. Impact of the Fecal Manipulation Procedure on Fecal Composition
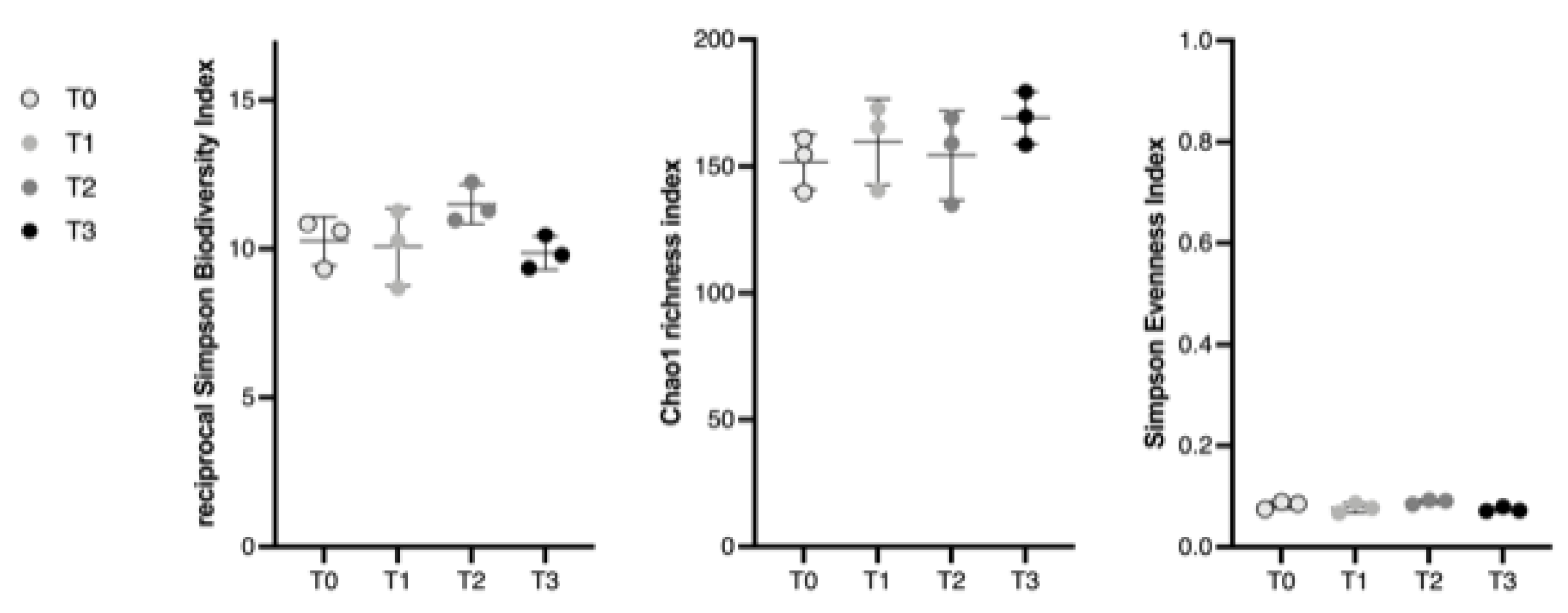
3.1.1. Alpha Diversity
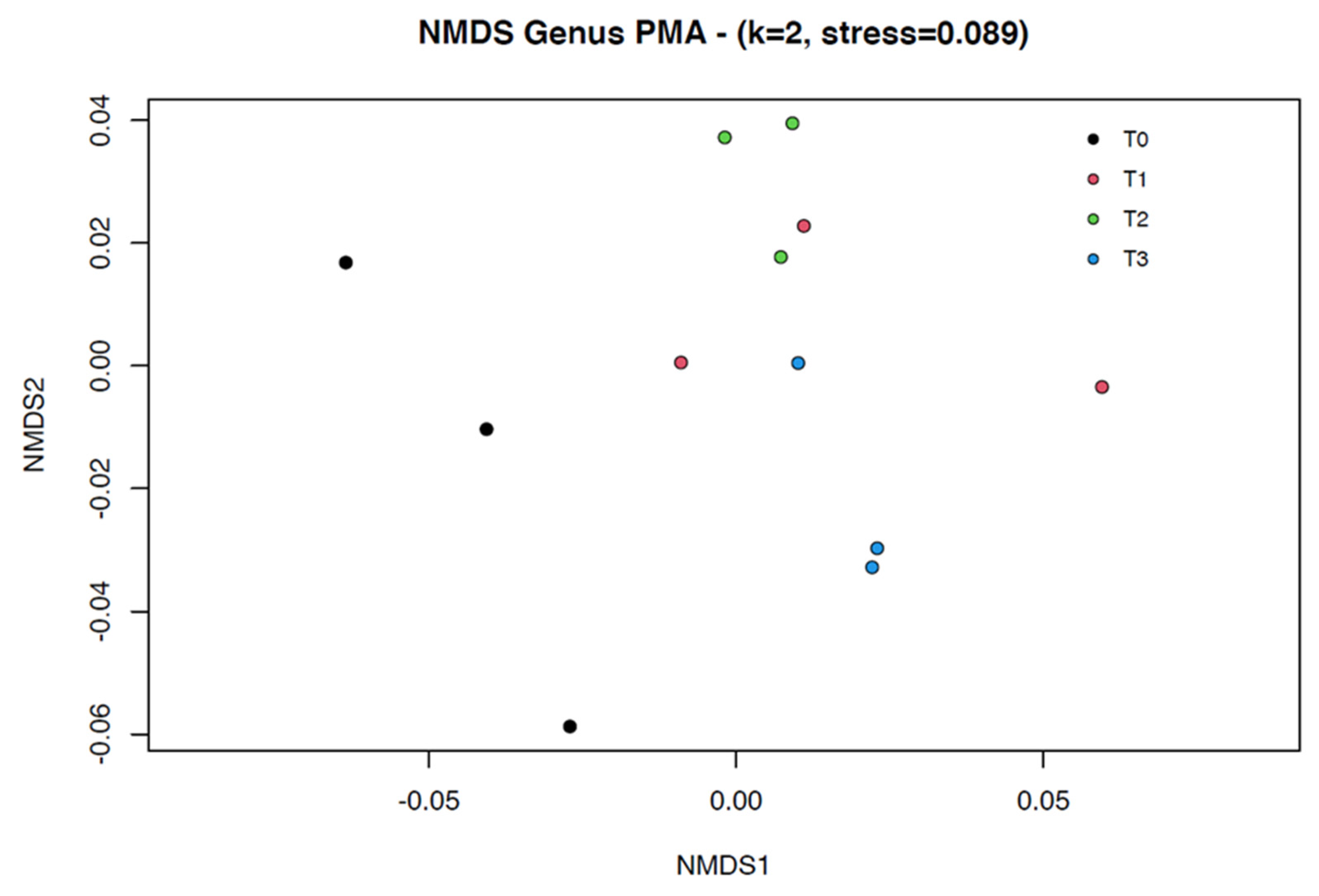
3.1.2. Beta Diversity
3.1.3. Major Bacterial Populations
3.2. Impact of the Fecal Manipulation Procedure on Fecal Bacterial Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilić-Stojanović, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European Consensus Conference on Faecal Microbiota Transplantation in Clinical Practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoster, A.; Weese, J.S.; Guardabassi, L. Probiotic Use in Horses—What Is the Evidence for Their Clinical Efficacy? J. Vet. Intern. Med. 2014, 28, 1640–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePeters, E.J.; George, L.W. Rumen Transfaunation. Immunol. Lett. 2014, 162, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.R.; Yasuda, K.; Divers, T.J.; Weese, J.S. Equine Faecal Microbiota Transplant: Current Knowledge, Proposed Guidelines and Future Directions. Equine Vet. Educ. 2018, 30, 151–160. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Kelly, C.R.; Mullish, B.H.; Allegretti, J.R.; Kassam, Z.; Putignani, L.; Fischer, M.; Keller, J.J.; Costello, S.P.; et al. International Consensus Conference on Stool Banking for Faecal Microbiota Transplantation in Clinical Practice. Gut 2019, 68, 2111–2121. [Google Scholar] [CrossRef] [Green Version]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A.Y.; et al. Durable Coexistence of Donor and Recipient Strains after Fecal Microbiota Transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef]
- Khoruts, A.; Sadowsky, M.J.; Hamilton, M.J. Development of Fecal Microbiota Transplantation Suitable for Mainstream Medicine. Clin. Gastroenterol. Hepatol. 2015, 13, 246–250. [Google Scholar] [CrossRef]
- van Nood, E.; Speelman, P.; Nieuwdorp, M.; Keller, J. Fecal Microbiota Transplantation. Curr. Opin. Gastroenterol. 2014, 30, 34–39. [Google Scholar] [CrossRef]
- Burz, S.D.; Abraham, A.-L.; Fonseca, F.; David, O.; Chapron, A.; Béguet-Crespel, F.; Cénard, S.; Roux, K.L.; Patrascu, O.; Levenez, F.; et al. A Guide for Ex Vivo Handling and Storage of Stool Samples Intended for Fecal Microbiota Transplantation. Sci. Rep. 2019, 9, 8897. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Wan, J.; Lu, Y.; Zhang, H.; Chen, X.; Su, Y.; Zhu, W. Active Bacterial Communities of Pig Fecal Microbiota Transplantation Suspension Prepared and Preserved under Different Conditions. AMB Express 2019, 9, 63. [Google Scholar] [CrossRef]
- Chu, N.D.; Smith, M.B.; Perrotta, A.R.; Kassam, Z.; Alm, E.J. Profiling Living Bacteria Informs Preparation of Fecal Microbiota Transplantations. PLoS ONE 2017, 12, e0170922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papanicolas, L.E.; Choo, J.M.; Wang, Y.; Leong, L.E.X.; Costello, S.P.; Gordon, D.L.; Wesselingh, S.L.; Rogers, G.B. Bacterial Viability in Faecal Transplants: Which Bacteria Survive? EBioMedicine 2019, 41, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, S.P.; Conlon, M.A.; Vuaran, M.S.; Roberts-Thomson, I.C.; Andrews, J.M. Faecal Microbiota Transplant for Recurrent Clostridium Difficile Infection Using Long-term Frozen Stool Is Effective: Clinical Efficacy and Bacterial Viability Data. Aliment. Pharmacol. Ther. 2015, 42, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Arai, K.; Asahara, T.; Takahashi, T.; Tsuji, H.; Matsumoto, S.; Takeuchi, I.; Kyodo, R.; Yamashiro, Y. Stool Preparation under Anaerobic Conditions Contributes to Retention of Obligate Anaerobes: Potential Improvement for Fecal Microbiota Transplantation. BMC Microbiol. 2021, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- Bellali, S.; Lagier, J.-C.; Raoult, D.; Khalil, J.B. Among Live and Dead Bacteria, the Optimization of Sample Collection and Processing Remains Essential in Recovering Gut Microbiota Components. Front. Microbiol. 2019, 10, 1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopper, J.J.; Alexander, T.L.; Kogan, C.J.; Berreta, A.R.; Burbick, C.R. In Vitro Evaluation of the Effect of Storage at −20°C and Proximal Gastrointestinal Conditions on Viability of Equine Fecal Microbiota Transplant. J. Equine Vet. Sci. 2020, 98, 103360. [Google Scholar] [CrossRef]
- Fouhy, F.; Deane, J.; Rea, M.C.; O’Sullivan, Ó.; Ross, R.P.; O’Callaghan, G.; Plant, B.J.; Stanton, C. The Effects of Freezing on Faecal Microbiota as Determined Using MiSeq Sequencing and Culture-Based Investigations. PLoS ONE 2015, 10, e0119355. [Google Scholar] [CrossRef]
- Papanicolas, L.E.; Wang, Y.; Choo, J.M.; Gordon, D.L.; Wesselingh, S.L.; Rogers, G.B. Optimisation of a Propidium Monoazide Based Method to Determine the Viability of Microbes in Faecal Slurries for Transplantation. J. Microbiol. Methods 2019, 156, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Nocker, A.; Sossa-Fernandez, P.; Burr, M.D.; Camper, A.K. Use of Propidium Monoazide for Live/Dead Distinction in Microbial Ecology. Appl. Environ. Microbiol. 2007, 73, 5111–5117. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.L.; Pitta, D.; Indugu, N.; Vecchiarelli, B.; Engiles, J.B.; Southwood, L.L. Characterization of the Fecal Microbiota of Healthy Horses. Am. J. Vet. Res. 2018, 79, 811–819. [Google Scholar] [CrossRef]
- Ngo, J.; Taminiau, B.; Fall, P.A.; Daube, G.; Fontaine, J. Ear Canal Microbiota—A Comparison between Healthy Dogs and Atopic Dogs without Clinical Signs of Otitis Externa. Vet. Dermatol. 2018, 29, 425-e140. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Taminiau, B.; de Lusancay, A.H.; Lecoq, L.; Amory, H.; Daube, G.; Cesarini, C. Effect of Oral Administration of Omeprazole on the Microbiota of the Gastric Glandular Mucosa and Feces of Healthy Horses. J. Vet. Intern. Med. 2020, 34, 2727–2737. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D. Evaluating Different Approaches That Test Whether Microbial Communities Have the Same Structure. ISME J. 2008, 2, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fastrès, A.; Taminiau, B.; Vangrinsven, E.; Tutunaru, A.-C.; Moyse, E.; Farnir, F.; Daube, G.; Clercx, C. Effect of an Antimicrobial Drug on Lung Microbiota in Healthy Dogs. Heliyon 2019, 5, e02802. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.C.; Silva, G.; Ramos, R.V.; Staempfli, H.R.; Arroyo, L.G.; Kim, P.; Weese, J.S. Characterization and Comparison of the Bacterial Microbiota in Different Gastrointestinal Tract Compartments in Horses. Vet. J. 2015, 205, 74–80. [Google Scholar] [CrossRef]
- Kauter, A.; Epping, L.; Semmler, T.; Antao, E.-M.; Kannapin, D.; Stoeckle, S.D.; Gehlen, H.; Lübke-Becker, A.; Günther, S.; Wieler, L.H.; et al. The Gut Microbiome of Horses: Current Research on Equine Enteral Microbiota and Future Perspectives. Anim. Microbiome 2019, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Satokari, R.; Mattila, E.; Kainulainen, V.; Arkkila, P.E.T. Simple Faecal Preparation and Efficacy of Frozen Inoculum in Faecal Microbiota Transplantation for Recurrent Clostridium Difficile Infection—An Observational Cohort Study. Aliment. Pharmacol. Ther. 2015, 41, 46–53. [Google Scholar] [CrossRef]
- Khanna, S. Microbiota Replacement Therapies: Innovation in Gastrointestinal Care. Clin. Pharmacol. Ther. 2017, 103, 102–111. [Google Scholar] [CrossRef]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘Unculturable’ Human Microbiota Reveals Novel Taxa and Extensive Sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullish, B.H.; Quraishi, M.N.; Segal, J.P.; McCune, V.L.; Baxter, M.; Marsden, G.L.; Moore, D.J.; Colville, A.; Bhala, N.; Iqbal, T.H.; et al. The Use of Faecal Microbiota Transplant as Treatment for Recurrent or Refractory Clostridium Difficile Infection and Other Potential Indications: Joint British Society of Gastroenterology (BSG) and Healthcare Infection Society (HIS) Guidelines. Gut 2018, 67, 1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low Counts of Faecalibacterium Prausnitzii in Colitis Microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Rajilic, M.; Biagi, E.; Tims, S.; Vos, W.M.D. Global and Deep Molecular Analysis of Microbiota Signatures in Fecal Samples from Patients with Irritable Bowel Syndrome. Gastroenterology 2011, 141, 10. [Google Scholar]
- McKinney, C.A.; Bedenice, D.; Pacheco, A.P.; Oliveira, B.C.M.; Paradis, M.-R.; Mazan, M.; Widmer, G. Assessment of Clinical and Microbiota Responses to Fecal Microbial Transplantation in Adult Horses with Diarrhea. PLoS ONE 2021, 16, e0244381. [Google Scholar] [CrossRef]
- Hagey, J.V.; Laabs, M.; Maga, E.A.; DePeters, E.J. Rumen Sampling Methods Bias Bacterial Communities Observed. PLoS ONE 2022, 17, e0258176. [Google Scholar] [CrossRef]
- Ren, Q.; Si, H.; Yan, X.; Liu, C.; Ding, L.; Long, R.; Li, Z.; Qiu, Q. Bacterial Communities in the Solid, Liquid, Dorsal, and Ventral Epithelium Fractions of Yak (Bos Grunniens) Rumen. MicrobiologyOpen 2020, 9, e963. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A Heritable Subset of the Core Rumen Microbiome Dictates Dairy Cow Productivity and Emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [Green Version]
- Bahl, M.I.; Bergström, A.; Licht, T.R. Freezing Fecal Samples Prior to DNA Extraction Affects the Firmicutes to Bacteroidetes Ratio Determined by Downstream Quantitative PCR Analysis. FEMS Microbiol. Lett. 2012, 329, 193–197. [Google Scholar] [CrossRef]
- Fittipaldi, M.; Nocker, A.; Codony, F. Progress in Understanding Preferential Detection of Live Cells Using Viability Dyes in Combination with DNA Amplification. J. Microbiol. Methods 2012, 91, 276–289. [Google Scholar] [CrossRef]
- de Bustamante, M.M.; Plummer, C.; MacNicol, J.; Gomez, D. Impact of Ambient Temperature Sample Storage on the Equine Fecal Microbiota. Animals 2021, 11, 819. [Google Scholar] [CrossRef] [PubMed]
- Schoster, A.; Mosing, M.; Jalali, M.; Staempfli, H.R.; Weese, J.S. Effects of Transport, Fasting and Anaesthesia on the Faecal Microbiota of Healthy Adult Horses. Equine Vet. J. 2015, 48, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Theelen, M.J.P.; Luiken, R.E.C.; Wagenaar, J.A.; van Oldruitenborgh-Oosterbaan, M.M.S.; Rossen, J.W.A.; Zomer, A.L. The Equine Faecal Microbiota of Healthy Horses and Ponies in The Netherlands: Impact of Host and Environmental Factors. Animals 2021, 11, 1762. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loublier, C.; Taminiau, B.; Heinen, J.; Lecoq, L.; Amory, H.; Daube, G.; Cesarini, C. Evaluation of Bacterial Composition and Viability of Equine Feces after Processing for Transplantation. Microorganisms 2023, 11, 231. https://doi.org/10.3390/microorganisms11020231
Loublier C, Taminiau B, Heinen J, Lecoq L, Amory H, Daube G, Cesarini C. Evaluation of Bacterial Composition and Viability of Equine Feces after Processing for Transplantation. Microorganisms. 2023; 11(2):231. https://doi.org/10.3390/microorganisms11020231
Chicago/Turabian StyleLoublier, Clémence, Bernard Taminiau, Julia Heinen, Laureline Lecoq, Hélène Amory, Georges Daube, and Carla Cesarini. 2023. "Evaluation of Bacterial Composition and Viability of Equine Feces after Processing for Transplantation" Microorganisms 11, no. 2: 231. https://doi.org/10.3390/microorganisms11020231
APA StyleLoublier, C., Taminiau, B., Heinen, J., Lecoq, L., Amory, H., Daube, G., & Cesarini, C. (2023). Evaluation of Bacterial Composition and Viability of Equine Feces after Processing for Transplantation. Microorganisms, 11(2), 231. https://doi.org/10.3390/microorganisms11020231