
Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primer and Probe Design
2.2. 10 μM PMAxx-RPA Exo Assay
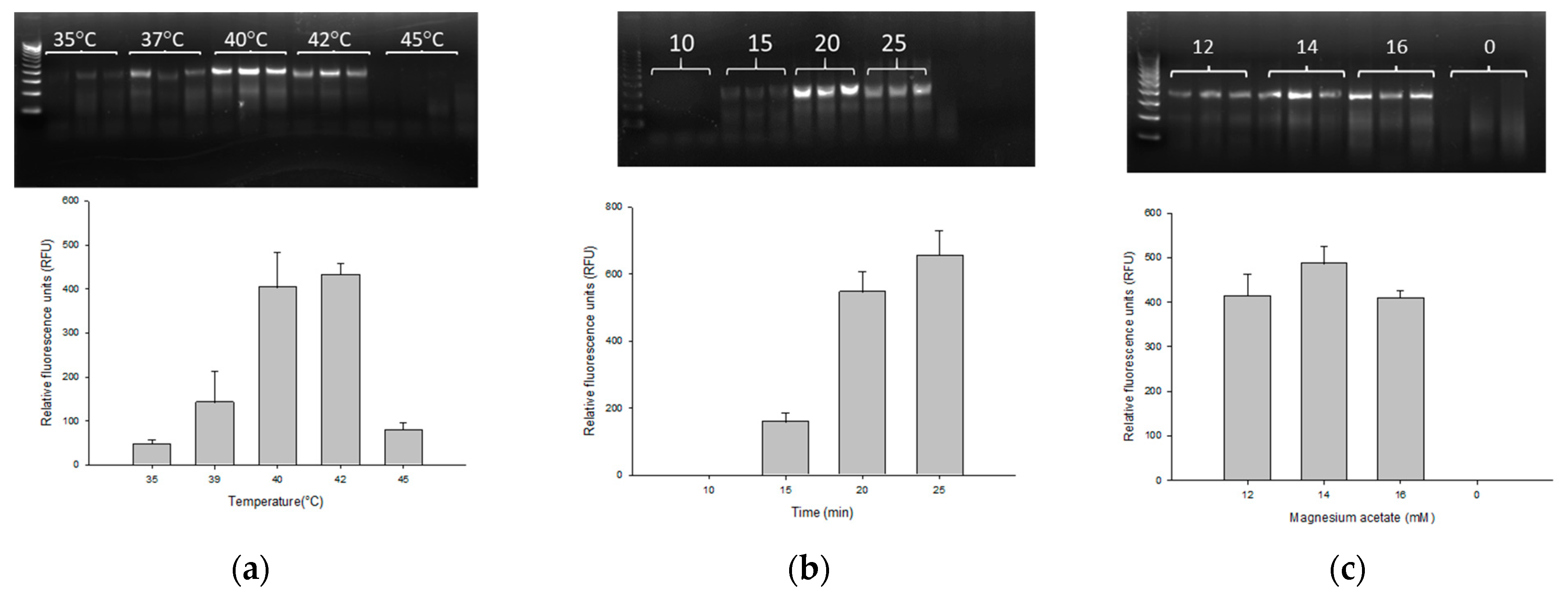
2.3. Optimization of Temperature, Reaction Time and Concentration of Magnesium Acetate for PMAxx-RPA Exo Assay
2.4. True-Negative Rate (Specificity) and Limit of Detection (LOD)
2.5. Effect of PMAxx-RPA Exo Assay in Presence of Antiseptics and Cell Lysates
2.5.1. Evaluation of Various Concentrations of CHX and BZK
2.5.2. Comparing DNA Extraction Methods Using the Boiling Method and a Commercial Kit
2.6. Detection of Live/Dead B. multivorans HI2229 in CHX and BZK Solutions
3. Results
3.1. Optimization of the PMAxx-RPA Exo Assay
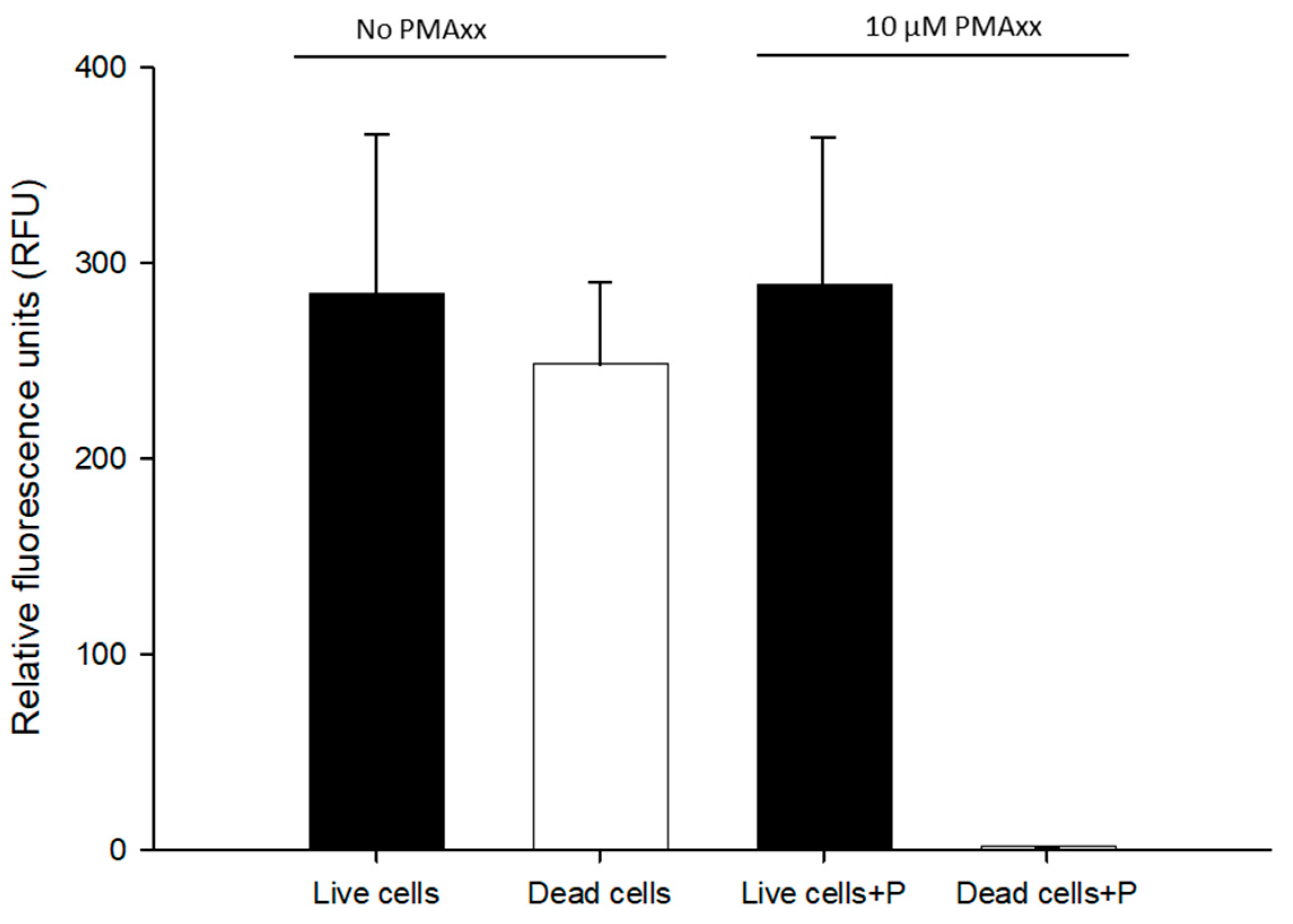
3.2. Evaluation of Live/Dead Cells with PMAxx Treatment
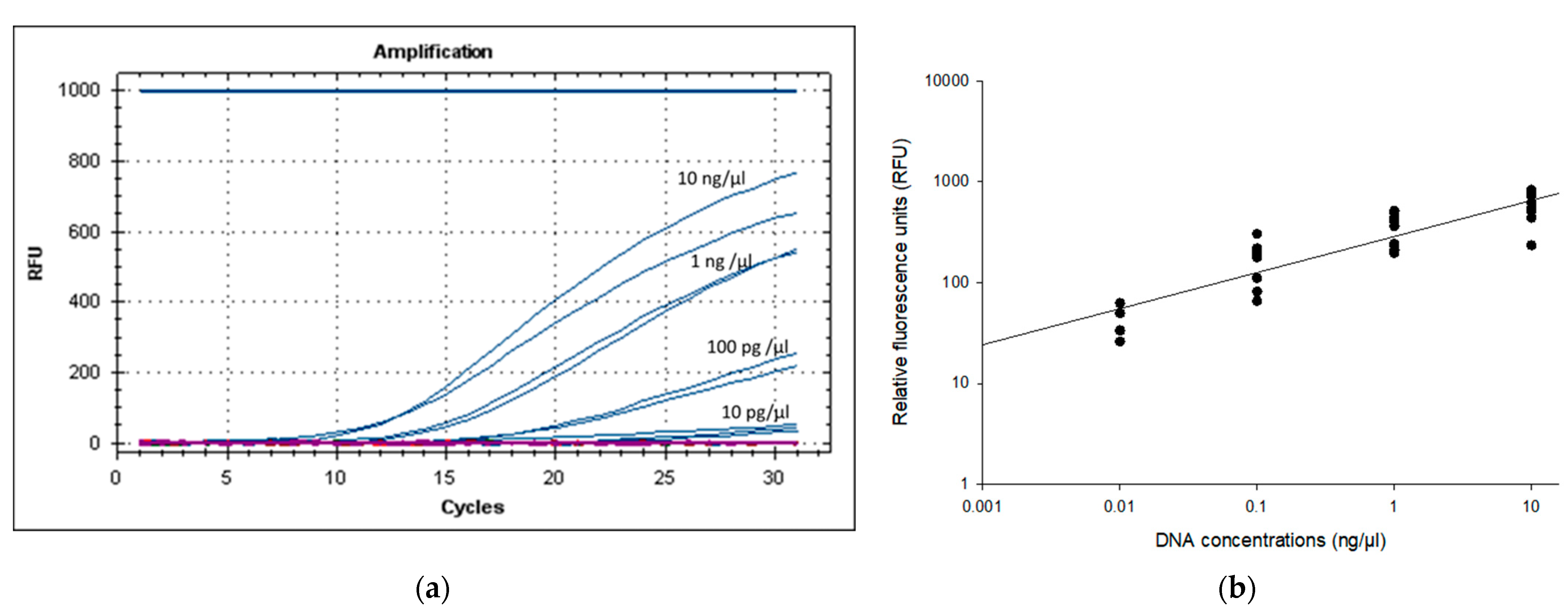
3.3. Specificity and LOD of PMAxx-RPA Exo Assay
3.4. Assessment of PMAxx-RPA Exo Conditions
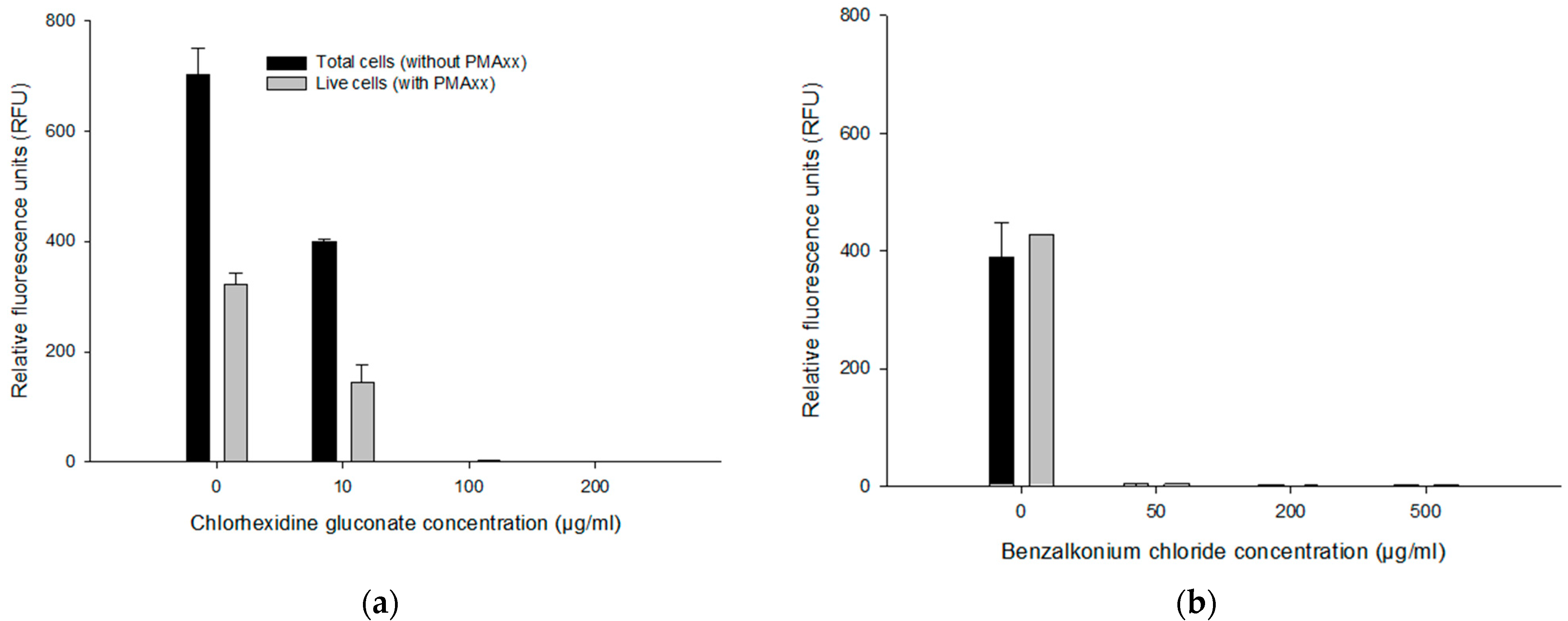
3.4.1. Effect of CHX and BZK on the PMAxx-RPA Exo Assay
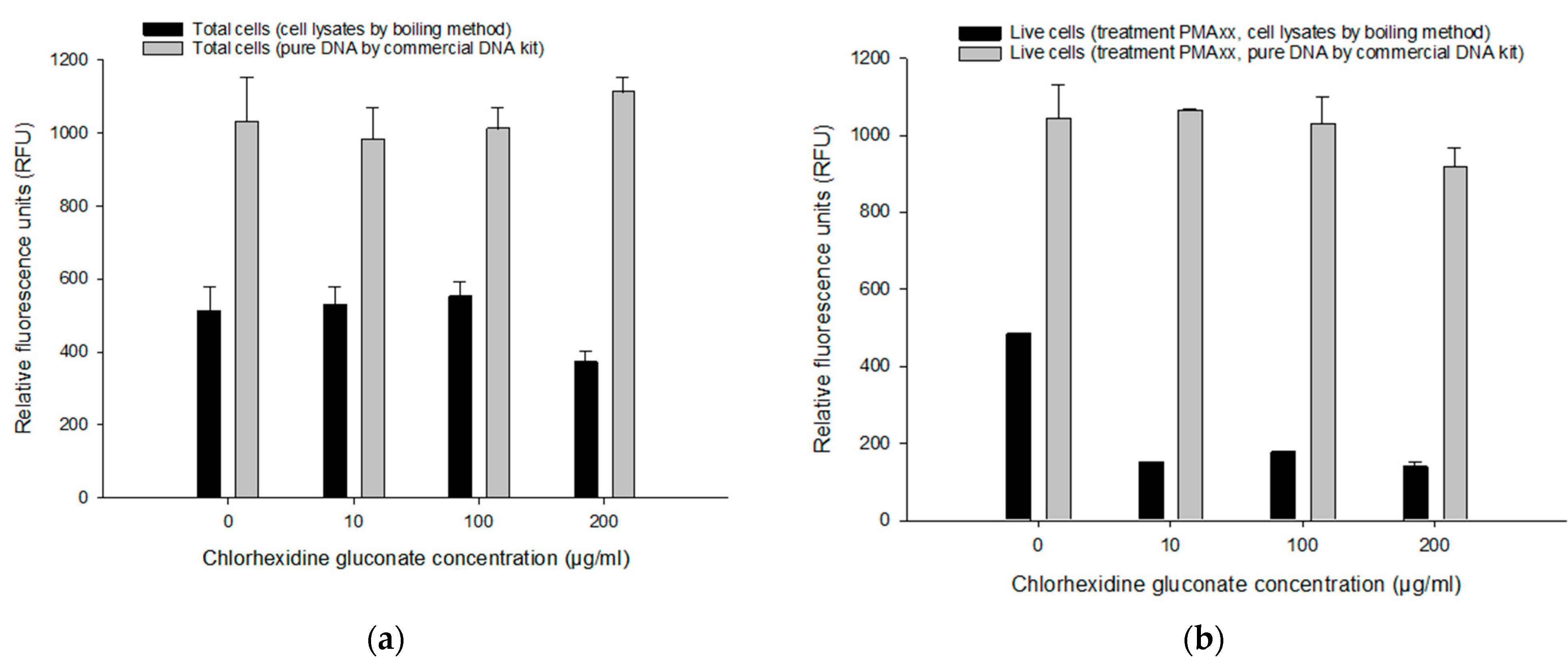
3.4.2. Comparing Pure DNA and Cell Lysates on the PMAxx-RPA Exo Assay
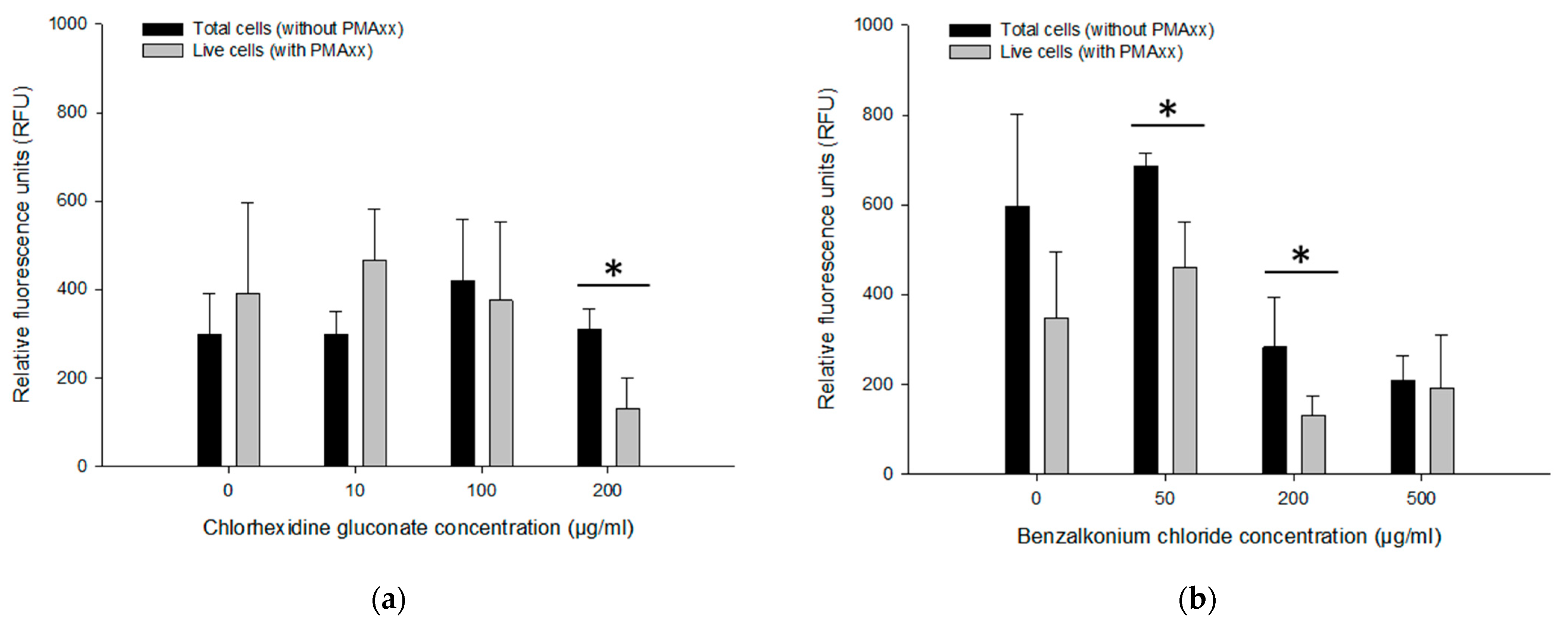
3.5. Assessing Live/Dead Cells in CHX and BZK Solutions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Depoorter, E.; De Canck, E.; Peeters, C.; Wieme, A.D.; Cnockaert, M.; Zlosnik, J.E.A.; LiPuma, J.J.; Coenye, T.; Vandamme, P. Burkholderia cepacia complex taxon K: Where to split? Front. Microbiol. 2020, 11, 1594. [Google Scholar] [CrossRef]
- Tavares, M.; Kozak, M.; Balola, A.; Sa-Correia, I. Burkholderia cepacia complex bacteria: A feared contamination risk in water-based pharmaceutical products. Clin. Microbiol. Rev. 2020, 33, e00139-19. [Google Scholar] [CrossRef] [PubMed]
- Scoffone, V.C.; Trespidi, G.; Barbieri, G.; Irudal, S.; Israyilova, A.; Buroni, S. Methodological tools to study species of the genus Burkholderia. Appl. Microbiol. Biotechnol. 2021, 105, 9019–9034. [Google Scholar] [CrossRef]
- Angrup, A.; Kanaujia, R.; Biswal, M.; Ray, P. Systematic review of ultrasound gel associated Burkholderia cepacia complex outbreaks: Clinical presentation, sources and control of outbreak. Am. J. Infect. Control 2022, 50, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L. Analysis of FDA Enforcement Reports (2012–2019) to Determine the Microbial Diversity in Contaminated Non-Sterile and Sterile Drugs. Am. Pharm. Rev. 2019, 4, 1–21. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/518912-Analysis-of-FDA-Enforcement-Reports-2012-2019-to-Determine-the-Microbial-Diversity-in-Contaminated-Non-Sterile-and-Sterile-Drugs/ (accessed on 3 April 2023).
- USP. <60> Microbiological Examination of Nonsterile Products—Tests for Burkholderia cepacia Complex. 2018. Available online: http://www.usppf.com/pf/pub/data/v445/CHA_IPR_445_c60.xml (accessed on 17 January 2023).
- Ahn, Y.; Gibson, B.; Williams, A.; Alusta, P.; Buzatu, D.A.; Lee, Y.J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Cerniglia, C.E. A comparison of culture-based, real-time PCR, droplet digital PCR and flow cytometric methods for the detection of Burkholderia cepacia complex in nuclease-free water and antiseptics. J. Ind. Microbiol. Biotechnol. 2020, 47, 475–484. [Google Scholar] [CrossRef]
- Jimenez, L.; Jashari, T.; Vasquez, J.; Zapata, S.; Bochis, J.; Kulko, M.; Ellman, V.; Gardner, M.; Choe, T. Real-Time PCR detection of Burkholderia cepacia in pharmaceutical products contaminated with low levels of bacterial contamination. PDA J. Pharm. Sci. Technol. 2018, 72, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Kweon, O.; Lee, Y.-J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Ahn, Y. Loop-mediated isothermal amplification (LAMP) assay for detecting Burkholderia cepacia complex in non-sterile pharmaceutical products. Pathogens 2021, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zheng, X.; Kan, B.; Li, W.; Zhang, W.; Jiang, T.; Lu, J.; Qin, A. Rapid detection of Burkholderia pseudomallei with a lateral flow recombinase polymerase amplification assay. PLoS ONE 2019, 14, e0213416. [Google Scholar] [CrossRef]
- Li, J.; Zhong, Q.; Shang, M.Y.; Li, M.; Jiang, Y.S.; Zou, J.J.; Ma, S.S.; Huang, Q.; Lu, W.P. Preliminary evaluation of rapid visual identification of Burkholderia pseudomallei using a newly developed lateral flow strip-based recombinase polymerase amplification (LF-RPA) system. Front. Cell Infect. Microbiol. 2021, 11, 804737. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Pal, V.; Tripathi, N.K.; Goel, A.K. A recombinase polymerase amplification lateral flow assay for rapid detection of Burkholderia pseudomallei, the causative agent of melioidosis. Braz. J. Microbiol. 2022, 53, 185–193. [Google Scholar] [CrossRef]
- Saxena, A.; Pal, V.; Tripathi, N.K.; Goel, A.K. Development of a rapid and sensitive recombinase polymerase amplification-lateral flow assay for detection of Burkholderia mallei. Transbound. Emerg. Dis. 2019, 66, 1016–1022. [Google Scholar] [CrossRef]
- Fu, H.; Gan, L.; Tian, Z.; Han, J.; Du, B.; Xue, G.; Feng, Y.; Zhao, H.; Cui, J.; Yan, C.; et al. Rapid detection of Burkholderia cepacia complex carrying the 16S rRNA gene in clinical specimens by recombinase-aided amplification. Front. Cell. Infect. Microbiol. 2022, 12, 984140. [Google Scholar] [CrossRef]
- Wong, M.Y.; Tseng, Y.H.; Huang, T.Y.; Lin, B.S.; Tung, C.W.; Chu, C.S.; Huang, Y.K. Comparison of microbiological characteristics and genetic diversity between Burkholderia cepacia complex isolates from vascular access and other clinical infections. Microorganisms 2021, 9, 51. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Kweon, O.; Lee, Y.J.; Hussong, D.; Marasa, B.; Ahn, Y. A Propidium monoazide (PMAxx)-droplet digital PCR (ddPCR) for the detection of viable Burkholderia cepacia complex in nuclease-Free water and antiseptics. Microorganisms 2022, 10, 943. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Williams, A.; Le, D.; Kweon, O.; Alusta, P.; Buzatu, D.A.; Ahn, Y. Specific detection and enumeration of Burkholderia cepacia complex by flow cytometry using a fluorescence-labeled oligonucleotide probe. Microorganisms 2022, 10, 1170. [Google Scholar] [CrossRef]
- Higgins, M.; Ravenhall, M.; Ward, D.; Phelan, J.; Ibrahim, A.; Forrest, M.S.; Clark, T.G.; Campino, S. PrimedRPA: Primer design for recombinase polymerase amplification assays. Bioinformatics 2019, 35, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Georgoutsou-Spyridonos, M.; Filippidou, M.; Kaprou, G.D.; Mastellos, D.C.; Chatzandroulis, S.; Tserepi, A. Isothermal recombinase polymerase amplification (RPA) of E. coli gDNA in commercially fabricated PCB-based microfluidic platforms. Micromachines 2021, 12, 1387. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Ahn, Y.; LiPuma, J.J.; Hussong, D.; Cerniglia, C.E. Survival and susceptibility of Burkholderia cepacia complex in chlorhexidine gluconate and benzalkonium chloride. J. Ind. Microbiol. Biotechnol. 2015, 42, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Furlan, J.P.R.; Pitondo-Silva, A.; Braz, V.S.; Gallo, I.F.L.; Stehling, E.G. Evaluation of different molecular and phenotypic methods for identification of environmental Burkholderia cepacia complex. World J. Microb. Biot. 2019, 35, 39. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, J.L.; Zhou, J.; Hu, M.D.; Zhang, Q.; Kong, N.; Ren, H.G.; Liang, L.; Yue, J.J. Genome-based classification of Burkholderia cepacia complex provides new insight into its taxonomic status. Biol. Direct 2020, 15, 6. [Google Scholar] [CrossRef]
- Martinucci, M.; Roscetto, E.; Iula, V.D.; Votsi, A.; Catania, M.R.; De Gregorio, E. Accurate identification of members of the Burkholderia cepacia complex in cystic fibrosis sputum. Lett. Appl. Microbiol. 2016, 62, 221–229. [Google Scholar] [CrossRef]
- Jordan, L.; Kurtz, R. Optical Design of CFX96™ Real-Time PCR Detection System Eliminates the Requirement of a Passive Reference Dye. 2010. Available online: https://www.bio-rad.com/en-us/applications-technologies/normalization-real-time-pcr-fluorescence-data-with-rox-passive-reference-dye?ID=MW472W15#Normalization_of_Fluorescence_Data_Using_ROX (accessed on 17 January 2023).
- Li, J.; Zhou, D.; Xie, G.; Deng, M.; Feng, X.; Xu, H. PMAxx combined with recombinase aided amplification technique for specific and rapid detection of Salmonella in milk. Food Anal. Methods 2022, 15, 1769–1777. [Google Scholar] [CrossRef]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar. J. 2014, 13, 99. [Google Scholar] [CrossRef]
- Liu, H.B.; Zang, Y.X.; Du, X.J.; Li, P.; Wang, S. Development of an isothermal amplification-based assay for the rapid visual detection of Salmonella bacteria. J. Dairy Sci. 2017, 100, 7016–7025. [Google Scholar] [CrossRef]
- Merk, S.; Meyer, H.; Greiser-Wilke, I.; Sprague, L.D.; Neubauer, H. Detection of Burkholderia cepacia DNA from artificially infected EDTA-blood and lung tissue comparing different DNA isolation methods. J. Vet. Med. B 2006, 53, 281–285. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, J.M.; Kweon, O.; Kim, S.J.; Jones, R.C.; Woodling, K.; Gamboa da Costa, G.; LiPuma, J.J.; Hussong, D.; Marasa, B.S.; et al. Intrinsic resistance of Burkholderia cepacia complex to benzalkonium chloride. MBio 2016, 7, e01716-16. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Bentham, R.H.; Ross, K.E. Limitations of using propidium monoazide with qPCR to discriminate between live and dead Legionella in biofilm samples. Microbiol. Insights 2014, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Exterkate, R.A.M.; Zaura, E.; Brandt, B.W.; Buijs, M.J.; Koopman, J.; Crielaard, W.; ten Cate, J.M. The effect of propidium monoazide treatment on the measured bacterial composition of clinical samples after the use of a mouthwash. Clin. Oral Investig. 2015, 19, 813–822. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/Probe | Sequence (5′–3′) | Primer Length (nt) |
|---|---|---|
| gbpT-F gbpT-R | ACGCTGTCGTCGACGATCATCAGCCTCGTGCT ACCATCGACAGCGCCATCATGATCGTCTGGTT | 32 32 |
| Probe | TCGGCCGCGTGCCGGGGATCCTGTCGACGG[FAM-dT]G[THF]-[BHQ1-dT]CTTCGCGATGCCGCC | 49 |
| Group | Species | Strain | Results |
|---|---|---|---|
| BCC | Burkholderia cepacia | PC783 | + |
| AU24442 | + | ||
| Burkholderia stabilis | AU23340 | + | |
| Burkholderia ambifaria | HI2468 | + | |
| Burkholderia anthina | HI2738 | + | |
| Burkholderia metallica | AU0553 | + | |
| AU16697 | + | ||
| Burkholderia contaminans | HI3429 | + | |
| AU24637 | + | ||
| Burkholderia diffusa | AU1075 | + | |
| Burkholderia arboris | ES0263a | + | |
| AU22095 | + | ||
| Burkholderia lata | HI4002 | + | |
| Burkholderia multivorans | HI2229 | + | |
| AU24571 | + | ||
| Burkholderia vietnamiensis | HI2212 | + | |
| AU24694 | + | ||
| Burkholderia cenocepacia | AU1054 | + | |
| AU0222 | + | ||
| AU19236 | + | ||
| HI2976 | + | ||
| HI2485 | + | ||
| J2315 | + | ||
| Non-BCC | Burkholderia glumae | AU6208 | + |
| AU12450 | + | ||
| Burkholderia gladioli | AU26454 | + | |
| AU29541 | + | ||
| AU30473 | + | ||
| AU16341 | + | ||
| Burkholderia concitans | AU12121 | – | |
| Burkholderia oklahomensis | ES0634 | + | |
| Burkholderia plantarii | AU9801 | + | |
| AU37486 | + | ||
| Burkholderia thailandensis | AU13555 | + | |
| AU36262 | + | ||
| Burkholderia tropica | AU15822 | – | |
| AU19944 | – | ||
| Burkholderia fungorum | AU18377 | – | |
| AU35949 | – | ||
| Non-Burkholderia | Caballeronia zhejiangensis | AU10475 | – |
| AU12096 | – | ||
| Enterococcus faecalis | ATCC29212 | – | |
| Enterococcus durans | ATCC6056 | – | |
| Proteus mirabilis | ATCC7002 | – | |
| Enterococcus faecium | ATCC35667 | – | |
| ATCC49624 | – | ||
| Bacillus subtilis | ATCC6051 | – | |
| Citrobacter freundii | ATCC8090 | – | |
| Pseudomonas aeruginosa | PAO1 | – | |
| ATCC27853 | – | ||
| Yersinia enterocolitica subsp. entrocolitica | ATCC27729 | – | |
| Shigella sonnei | ATCC9290 | – | |
| Lactobacillus salivarius subsp. salivarius | ATCC11741 | – | |
| Enterobacter aerogenes | ATCC13048 | – | |
| Klebsiella pneumoniae | ATCC13883 | – | |
| Candida albicans (Robin) Berkhout | ATCC10231 | – | |
| Salmonella enterica | isolates | – | |
| Paenibacillus lautus | isolates | – | |
| Brevibacillus laterosporus | isolates | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daddy Gaoh, S.; Kweon, O.; Ahn, Y. Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms 2023, 11, 1401. https://doi.org/10.3390/microorganisms11061401
Daddy Gaoh S, Kweon O, Ahn Y. Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms. 2023; 11(6):1401. https://doi.org/10.3390/microorganisms11061401
Chicago/Turabian StyleDaddy Gaoh, Soumana, Ohgew Kweon, and Youngbeom Ahn. 2023. "Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions" Microorganisms 11, no. 6: 1401. https://doi.org/10.3390/microorganisms11061401
APA StyleDaddy Gaoh, S., Kweon, O., & Ahn, Y. (2023). Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms, 11(6), 1401. https://doi.org/10.3390/microorganisms11061401