
Alpha-Gal Bound Aptamer and Vancomycin Synergistically Reduce Staphylococcus aureus Infection In Vivo



Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Bacterial Strains and Growth Conditions
2.3. In Vitro Cell Culture
2.4. Aptamer Stability
2.5. Bacterial Adhesion Assay
2.6. S. aureus Growth Inhibition with α-SA31
2.7. Phagocytosis Assay
2.8. Ethics Statement
2.9. Animals
2.10. In Vivo Accumulation of αSA31
2.11. α-gal Ab ELISA
2.12. Statistics
3. Results
3.1. Alphamer Characterization
3.2. 5′-Capping Did Not Affect Stability
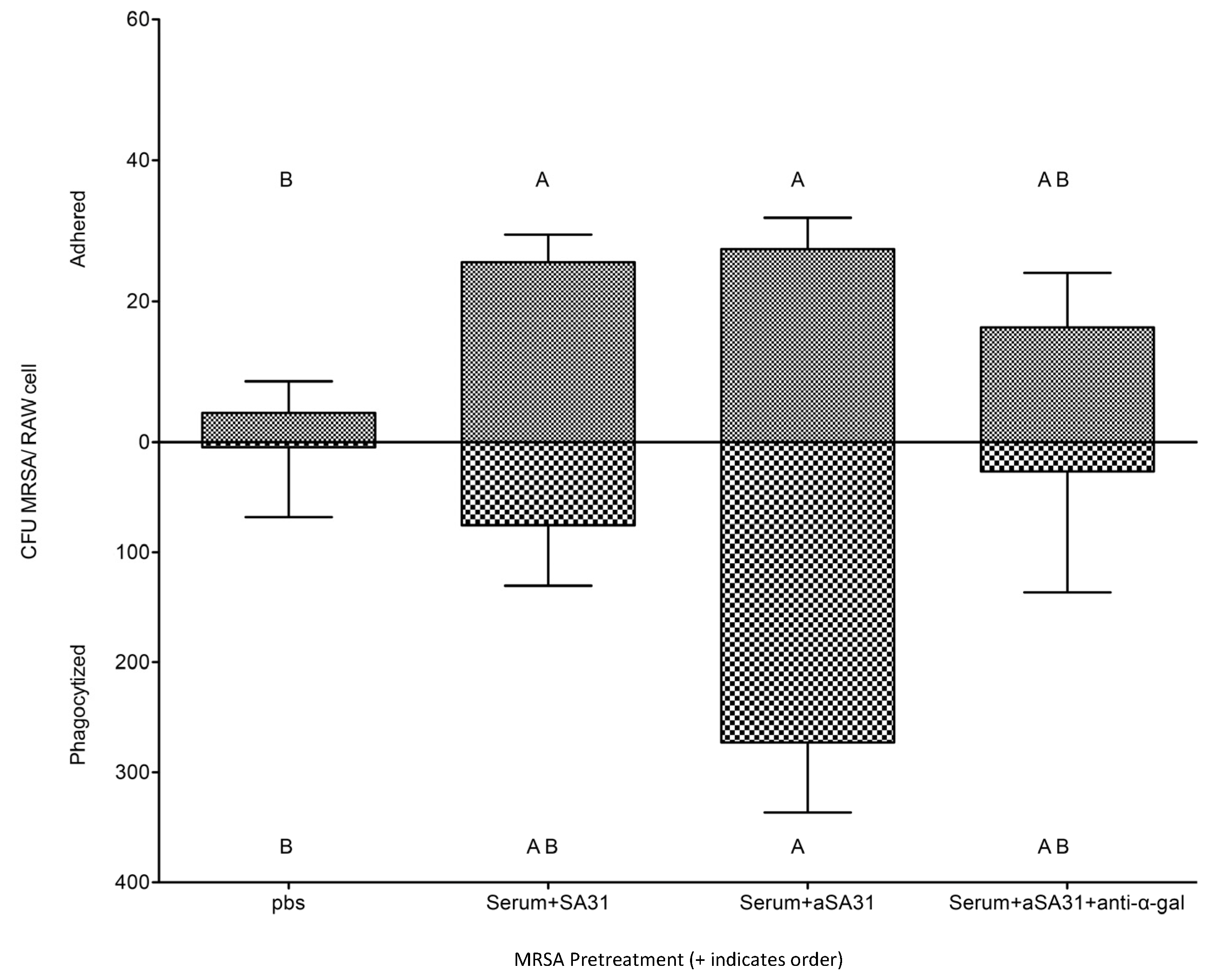
3.3. αSA31 Increased Phagocytosis In Vitro
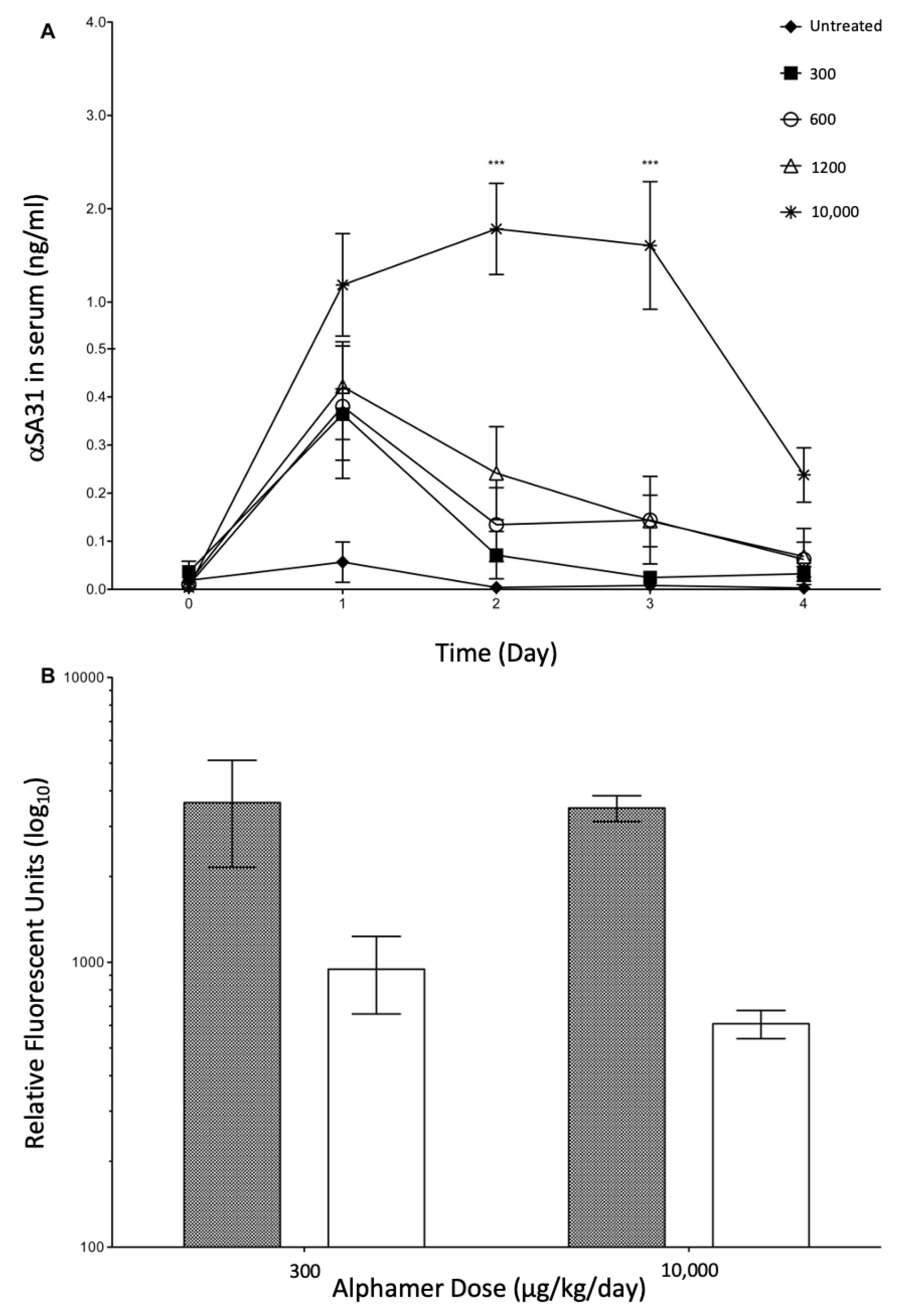
3.4. In Vivo Alphamer Accumulation
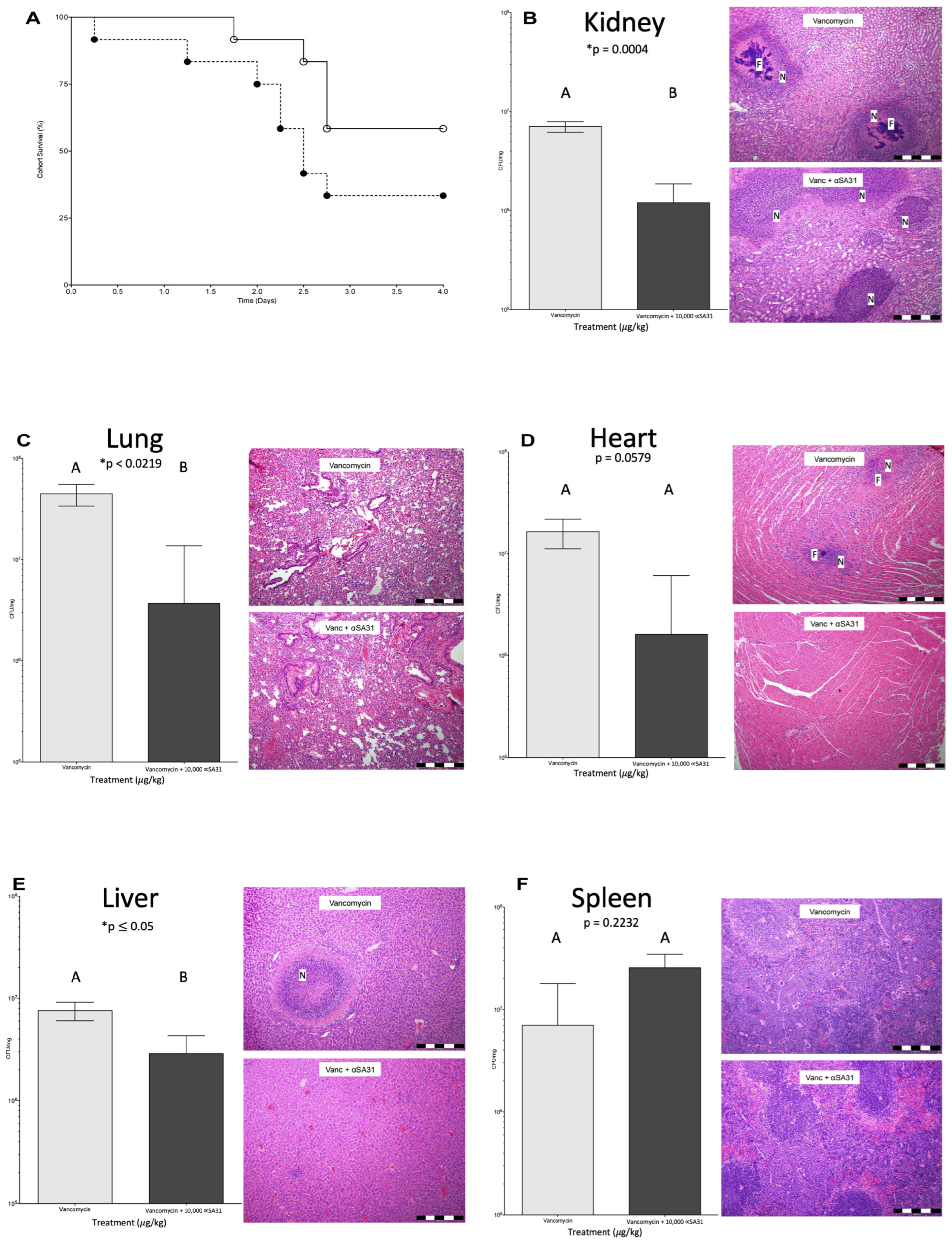
3.5. αSA31 Treatment during MRSA Sepsis
3.6. αSA31 Treatment Plus Vancomycin Resulted in Lower MRSA Organ Loads
3.7. Histology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Etter, D.; Corti, S.; Spirig, S.; Cernela, N.; Stephan, R.; Johler, S. Staphylococcus aureus Population Structure and Genomic Profiles in Asymptomatic Carriers in Switzerland. Front. Microbiol. 2020, 11, 1289. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Melles, D.C.; Nouwen, J.; van Leeuwen, W.B.; van Wamel, W.; Vos, M.C.; Wertheim, H.F.L.; Verbrugh, H.A. Co-evolutionary aspects of human colonisation and infection by Staphylococcus aureus. Infect. Genet. Evol. 2009, 9, 32–47. [Google Scholar] [PubMed]
- Lappin, E.; Ferguson, A.J. Gram-positive toxic shock syndromes. Lancet Infect. Dis. 2009, 9, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- Battisti, A.; Franco, A.; Merialdi, G.; Hasman, H.; Iurescia, M.; Lorenzetti, R.; Feltrin, F.; Zini, M.; Aarestrup, F. Heterogeneity among methicillin-resistant Staphylococcus aureus from Italian pig finishing holdings. Vet. Microbiol. 2010, 142, 361–366. [Google Scholar] [CrossRef]
- Guo, T.; Yang, Y.; Zhang, J.; Miao, Y.; Lin, F.; Zhu, S.; Zhang, C.; Wu, H. Ascorbate exacerbates iron toxicity on intestinal barrier function against Salmonella infection. J. Environ. Sci. Health Part C 2020, 38, 91–107. [Google Scholar] [CrossRef]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.B. In vitro activity of oritavancin against community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA), vancomycin-intermediate S. aureus (VISA), vancomycin-resistant S. aureus (VRSA) and daptomycin-non-susceptible S. aureus (DNSSA). Int. J. Antimicrob. Agents 2010, 36, 69–72. [Google Scholar]
- Sulaiman, J.E.; Wu, L.; Lam, H. Mutation in the Two-Component System Regulator YycH Leads to Daptomycin Tolerance in Methicillin-Resistant Staphylococcus aureus upon Evolution with a Population Bottleneck. Microbiol. Spectr. 2022, 10, e0168722. [Google Scholar] [CrossRef]
- Shoaib, M.; Aqib, A.I.; Muzammil, I.; Majeed, N.; Bhutta, Z.A.; Kulyar, M.F.-E.; Fatima, M.; Zaheer, C.-N.F.; Muneer, A.; Murtaza, M.; et al. MRSA compendium of epidemiology, transmission, pathophysiology, treatment, and prevention within one health framework. Front. Microbiol. 2023, 13, 1067284. [Google Scholar] [CrossRef]
- Alou, L.; Cafini, F.; Sevillano, D.; Unzueta, I.; Prieto, J. In vitro activity of mupirocin and amoxicillin-clavulanate alone and in combination against staphylococci including those resistant to methicillin. Int. J. Antimicrob. Agents 2004, 23, 513–516. [Google Scholar] [CrossRef]
- Gómez-Lus, M.L.; Prieto, J.; Gimenez, M.-J.; García, M.; Anta, L.; Aguilar, L. In vitro bactericidal activity of peak serum concentrations of co-amoxiclav, amoxicillin and vancomycin against methicillin-resistant Staphylococcus aureus. Rev. Esp. Quim. 1999, 12, 136–139. [Google Scholar]
- Moreillon, P. The efficacy of amoxycillin/clavulanate (Augmentin) in the treatment of severe staphylococcal infections. J. Chemother. 1994, 6 (Suppl. S2), 51–57. [Google Scholar] [PubMed]
- Gill, E.E.; Franco, O.L.; Hancock, R.E.W. Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Biol. Drug Des. 2014, 85, 56–78. [Google Scholar] [CrossRef] [Green Version]
- Sahu, M. Waging War against Extended Spectrum Beta Lactamase and Metallobetalactamase Producing Pathogens- Novel Adjuvant Antimicrobial Agent Cse1034- An Extended Hope. J. Clin. Diagn. Res. 2014, 8, DC20–DC23. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A. Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Galili, U. Anti-Gal: An abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology 2013, 140, 1–11. [Google Scholar] [CrossRef]
- Huai, G.; Qi, P.; Yang, H.; Wang, Y. Characteristics of α-Gal epitope, anti-Gal antibody, α1,3 galactosyltransferase and its clinical exploitation (Review). Int. J. Mol. Med. 2016, 37, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Galili, U. The α-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol. Cell Biol. 2005, 83, 674–686. [Google Scholar] [CrossRef]
- Posekany, K.J.; Pittman, H.K.; Bradfield, J.F.; Haisch, C.E.; Verbanac, K.M. Induction of cytolytic anti-Gal antibodies in alpha-1,3-galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect. Immun. 2002, 70, 6215–6222. [Google Scholar]
- Abdel-Motal, U.; Wang, S.; Lu, S.; Wigglesworth, K.; Galili, U. Increased immunogenicity of human immunodeficiency virus gp120 engineered to express Galalpha1-3Galbeta1-4GlcNAc-R epitopes. J. Virol. 2006, 80, 6943–6951. [Google Scholar]
- Duk, M.; Lisowska, E. Presence of natural anti-Galalpha1-4GalNAcbeta1-3Gal (anti-NOR) antibodies in animal sera. Glycoconj. J. 2006, 23, 585–590. [Google Scholar] [PubMed]
- Galili, U.; Mandrell, R.E.; Hamadeh, R.M.; Shohet, S.B.; Griffiss, J.M. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect. Immun. 1988, 56, 1730–1737. [Google Scholar] [PubMed]
- Hamadeh, R.M.; Jarvis, G.A.; Zhou, P.; Cotleur, A.C.; Griffiss, J.M. Bacterial enzymes can add galactose alpha 1,3 to human erythrocytes and creates a senescence-associated epitope. Infect. Immun. 1996, 64, 528–534. [Google Scholar]
- Lisowska, E.; Duk, M. Diversity of Natural Anti-α-Galactosyl Antibodies in Human Serum. Adv. Exp. Med. Biol. 2011, 705, 571–583. [Google Scholar] [CrossRef]
- Rodriguez, I.A.; Welsh, R.M. Possible Role of a Cell Surface Carbohydrate in Evolution of Resistance to Viral Infections in Old World Primates. J. Virol. 2013, 87, 8317–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernth Jensen, J.M.; Laursen, N.S.; Jensen, R.K.; Andersen, G.R.; Jensenius, J.C.; Sørensen, U.B.S.; Thiel, S. Complement activation by human IgG antibodies to galactose-alpha-1,3-galactose. Immunology 2020, 161, 66–79. [Google Scholar] [PubMed]
- Perdomo, M.F.; Levi, M.; Sällberg, M.; Vahlne, A. Neutralization of HIV-1 by redirection of natural antibodies. Proc. Natl. Acad. Sci. USA 2008, 105, 12515–12520. [Google Scholar] [CrossRef]
- Kanwar, J.R.; Roy, K.; Kanwar, R.K. Chimeric aptamers in cancer cell-targeted drug delivery. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 459–477. [Google Scholar] [CrossRef]
- Kristian, S.A.; Hwang, J.H.; Hall, B.; Leire, E.; Iacomini, J.; Old, R.; Galili, U.; Roberts, C.; Mullis, K.B.; Westby, M.; et al. Retargeting pre-existing human antibodies to a bacterial pathogen with an alpha-Gal conjugated aptamer. J. Mol. Med. 2015, 93, 619–631. [Google Scholar] [CrossRef] [Green Version]
- You, K.M.; Lee, S.H.; Im, A.; Lee, S.B. Aptamers as functional nucleic acids:In vitro selection and biotechnological applications. Biotechnol. Bioprocess Eng. 2003, 8, 64–75. [Google Scholar] [CrossRef]
- Cao, X.; Li, S.; Chen, L.; Ding, H.; Xu, H.; Huang, Y.; Li, J.; Liu, N.; Cao, W.; Zhu, Y.; et al. Combining use of a panel of ssDNA aptamers in the detection of Staphylococcus aureus. Nucleic Acids Res. 2009, 37, 4621–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwehand, A.C.; Salminen, S. In vitroAdhesion Assays for Probiotics and theirin vivoRelevance: A Review. Microb. Ecol. Health Dis. 2003, 15, 175–184. [Google Scholar] [CrossRef]
- Chen, P.; Reiter, T.; Huang, B.; Kong, N.; Weimer, B.C. Prebiotic Oligosaccharides Potentiate Host Protective Responses against L. Monocytogenes Infection. Pathogens 2017, 6, 68. [Google Scholar]
- Arabyan, N.; Park, D.; Foutouhi, S.; Weis, A.M.; Huang, B.C.; Williams, C.C.; Desai, P.; Shah, J.; Jeannotte, R.; Kong, N.; et al. Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling. Sci. Rep. 2016, 6, 29525. [Google Scholar] [CrossRef]
- De Ridder, L.; Mareel, M.; Vakaet, L. Adhesion of malignant and nonmalignant cells to cultured embryonic substrates. Cancer Res 1975, 35, 3164–3171. [Google Scholar]
- Lominadze, D.G.; Saari, J.T.; Miller, F.N.; Catalfamo, J.L.; E Justus, D.; A Schuschke, D. Platelet aggregation and adhesion during dietary copper deficiency in rats. Thromb. Haemost. 1996, 75, 630–634. [Google Scholar]
- Elsinghorst, E.A. Measurement of invasion by gentamicin resistance. Methods Enzymol. 1994, 236, 405–420. [Google Scholar] [CrossRef]
- Francois, P.; Pittet, D.; Bento, M.; Pepey, B.; Vaudaux, P.; Lew, D.; Schrenzel, J. Rapid Detection of Methicillin-Resistant Staphylococcus aureus Directly from Sterile or Nonsterile Clinical Samples by a New Molecular Assay. J. Clin. Microbiol. 2003, 41, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Nadkarni, M.A.; Martin, F.E.; Jacques, N.A.; Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 2002, 148, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Dodge, J.T.; Mitchell, C.; Hanahan, D.J. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 1963, 100, 119–130. [Google Scholar] [CrossRef]
- Jeon, S.H.; Kayhan, B.; Ben-Yedidia, T.; Arnon, R. A DNA Aptamer Prevents Influenza Infection by Blocking the Receptor Binding Region of the Viral Hemagglutinin. J. Biol. Chem. 2004, 279, 48410–48419. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Rao, N.; Lai, K.-T.A.; Gu, Y.; Sun, S.; Fuchs, A.; Fung-Leung, W.-P.; Colonna, M.; Karlsson, L. CpG-induced tyrosine phosphorylation occurs via a TLR9-independent mechanism and is required for cytokine secretion. J. Cell Biol. 2006, 172, 1057–1068. [Google Scholar]
- Galili, U.; LaTemple, D.C.; Radic, M.Z. A Sensitive Assay for Measuring α-gal epitope expression on cells by a monoclonal anti-gal antibody1. Transplantation 1998, 65, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Katakura, T.; Yoshida, T.; Kobayashi, M.; Herndon, D.N.; Suzuki, F. Immunological control of methicillin-resistant Staphylococcus aureus (MRSA) infection in an immunodeficient murine model of thermal injuries. Clin. Exp. Immunol. 2005, 142, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Miyazaki, H.; Ono, S.; Inatsu, A.; Nakashima, H.; Tsujimoto, H.; Shinomiya, N.; Saitoh, D.; Seki, S. Enhancement of Neutrophil Function by Interleukin-18 Therapy Protects Burn-Injured Mice from Methicillin-Resistant Staphylococcus aureus. Infect. Immun. 2011, 79, 2670–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin as a treatment for systemic Staphylococcus aureus infection in a mouse model. J. Antimicrob. Chemother. 2007, 60, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.-M.; Hsu, C.-C.; Yin, M.-C. Meticillin-resistant Staphylococcus aureus infection in diabetic mice enhanced inflammation and coagulation. J. Med. Microbiol. 2006, 55, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Kambe, F.; Lu, X.; Kobayashi, N.; Ohmori, S.; Seo, H. Glutathionylation of two cysteine residues in paired domain regulates DNA binding activity of Pax-8. J. Biol. Chem. 2005, 280, 25901–25906. [Google Scholar]
- Dasari, P.; Nordengrün, M.; Vilhena, C.; Steil, L.; Abdurrahman, G.; Surmann, K.; Dhople, V.; Lahrberg, J.; Bachert, C.; Skerka, C.; et al. The Protease SplB of Staphylococcus aureus Targets Host Complement Components and Inhibits Complement-Mediated Bacterial Opsonophagocytosis. J. Bacteriol. 2022, 204, e0018421. [Google Scholar] [CrossRef]
- Laarman, A.; Milder, F.; van Strijp, J.; Rooijakkers, S. Complement inhibition by gram-positive pathogens: Molecular mechanisms and therapeutic implications. J. Mol. Med. 2010, 88, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Agramonte-Hevia, J.; González-Arenas, A.; Barrera, D.; Velasco-Velázquez, M. Gram-negative bacteria and phagocytic cell interaction mediated by complement receptor 3. FEMS Immunol. Med. Microbiol. 2002, 34, 255–266. [Google Scholar]
- Top, E.A.V.; A Perry, G.; Gentry-Nielsen, M.J. A novel flow cytometric assay for measurement of In Vivo pulmonary neutrophil phagocytosis. BMC Microbiol. 2006, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- van Kessel, K.P.; Bestebroer, J.; van Strijp, J.A. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [PubMed] [Green Version]
- O’Riordan, K.; Lee, J.C. Staphylococcus aureus Capsular Polysaccharides. Clin. Microbiol. Rev. 2004, 17, 218–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijh, P.C.; Barselaar, M.T.V.D.; Daha, M.R.; Van Furth, R. Participation of immunoglobulins and complement components in the intracellular killing of Staphylococcus aureus and Escherichia coli by human granulocytes. Infect. Immun. 1981, 33, 714–724. [Google Scholar]
- Peterson, P.K.; Wilkinson, B.J.; Kim, Y.; Schmeling, D.; Quie, P.G. Influence of encapsulation on staphylococcal opsonization and phagocytosis by human polymorphonuclear leukocytes. Infect. Immun. 1978, 19, 943–949. [Google Scholar]
- Verhoef, J.; Peterson, P.; Kim, Y.; Sabath, L.D.; Quie, P.G. Opsonic requirements for staphylococcal phagocytosis. Heterogeneity among strains. Immunology 1977, 33, 191–197. [Google Scholar]
- Rigby, K.M.; DeLeo, F.R. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 2011, 34, 237–259. [Google Scholar] [CrossRef] [Green Version]
- Chavanet, P. The ZEPHyR study: A randomized comparison of linezolid and vancomycin for MRSA pneumonia. Med. Et Mal. Infect. 2013, 43, 451–455. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Totsuka, K.; Shiseki, M.; Kikuchi, K.; Matsui, Y. Combined effects of vancomycin and imipenem against methicillin-resistant Staphylococcus aureus (MRSA) in vitro and in vivo. J. Antimicrob. Chemother. 1999, 44, 455–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [PubMed] [Green Version]
- Skaar, P.E.; Schneewind, O. Iron-regulated surface determinants (Isd) of Staphylococcus aureus: Stealing iron from heme. Microbes Infect. 2004, 6, 390–397. [Google Scholar] [PubMed]
- Torres, V.J.; Pishchany, G.; Humayun, M.; Schneewind, O.; Skaar, E.P. Staphylococcus aureus IsdB Is a Hemoglobin Receptor Required for Heme Iron Utilization. J. Bacteriol. 2006, 188, 8421–8429. [Google Scholar] [CrossRef] [Green Version]
- Visai, L.; Yanagisawa, N.; Josefsson, E.; Tarkowski, A.; Pezzali, I.; Rooijakkers, S.H.M.; Foster, T.J.; Speziale, P. Immune evasion by Staphylococcus aureus conferred by iron-regulated surface determinant protein IsdH. Microbiology 2009, 155, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar]
- Cascioferro, S.; Carbone, D.; Parrino, B.; Pecoraro, C.; Giovannetti, E.; Cirrincione, G.; Diana, P. Therapeutic Strategies To Counteract Antibiotic Resistance in MRSA Biofilm-Associated Infections. Chemmedchem 2021, 16, 65–80. [Google Scholar] [CrossRef]
- Kareem, S.; Aljubori, S.; Ali, M. Novel determination of spa gene diversity and its molecular typing among Staphylococcus aureus Iraqi isolates obtained from different clinical samples. New Microbes New Infect. 2020, 34, 100653. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | |||
|---|---|---|---|
| Treatment | SA31 | αSA31NH2 | αSA31 |
| Water | 69.337 B | 10,000 *A | 10,000 *A |
| Endonuclease (DNase1) | 1.178 D | 1.689 D | 1.233 D |
| 3′->5′ Exonuclease (Exo1) | 5.014 C,D | 9.494 C,D | 4.082 C,D |
| 5′->3′ Exonuclease (T5) | 1.88 D | 1.761 D | 2.099 D |
| Human serum (homo sapiens) | 3.938 C,D | 17.618 C | 5.222 C,D |
| Mouse serum (mus musculus) | 1.484 D | 2.467 D | 1.789 D |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doherty, M.K.; Shaw, C.; Woods, L.; Weimer, B.C. Alpha-Gal Bound Aptamer and Vancomycin Synergistically Reduce Staphylococcus aureus Infection In Vivo. Microorganisms 2023, 11, 1776. https://doi.org/10.3390/microorganisms11071776
Doherty MK, Shaw C, Woods L, Weimer BC. Alpha-Gal Bound Aptamer and Vancomycin Synergistically Reduce Staphylococcus aureus Infection In Vivo. Microorganisms. 2023; 11(7):1776. https://doi.org/10.3390/microorganisms11071776
Chicago/Turabian StyleDoherty, Matthew K., Claire Shaw, Leslie Woods, and Bart C. Weimer. 2023. "Alpha-Gal Bound Aptamer and Vancomycin Synergistically Reduce Staphylococcus aureus Infection In Vivo" Microorganisms 11, no. 7: 1776. https://doi.org/10.3390/microorganisms11071776
APA StyleDoherty, M. K., Shaw, C., Woods, L., & Weimer, B. C. (2023). Alpha-Gal Bound Aptamer and Vancomycin Synergistically Reduce Staphylococcus aureus Infection In Vivo. Microorganisms, 11(7), 1776. https://doi.org/10.3390/microorganisms11071776