

Changes in Bulk and Rhizosphere Soil Microbial Diversity Communities of Native Quinoa Due to the Monocropping in the Peruvian Central Andes

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
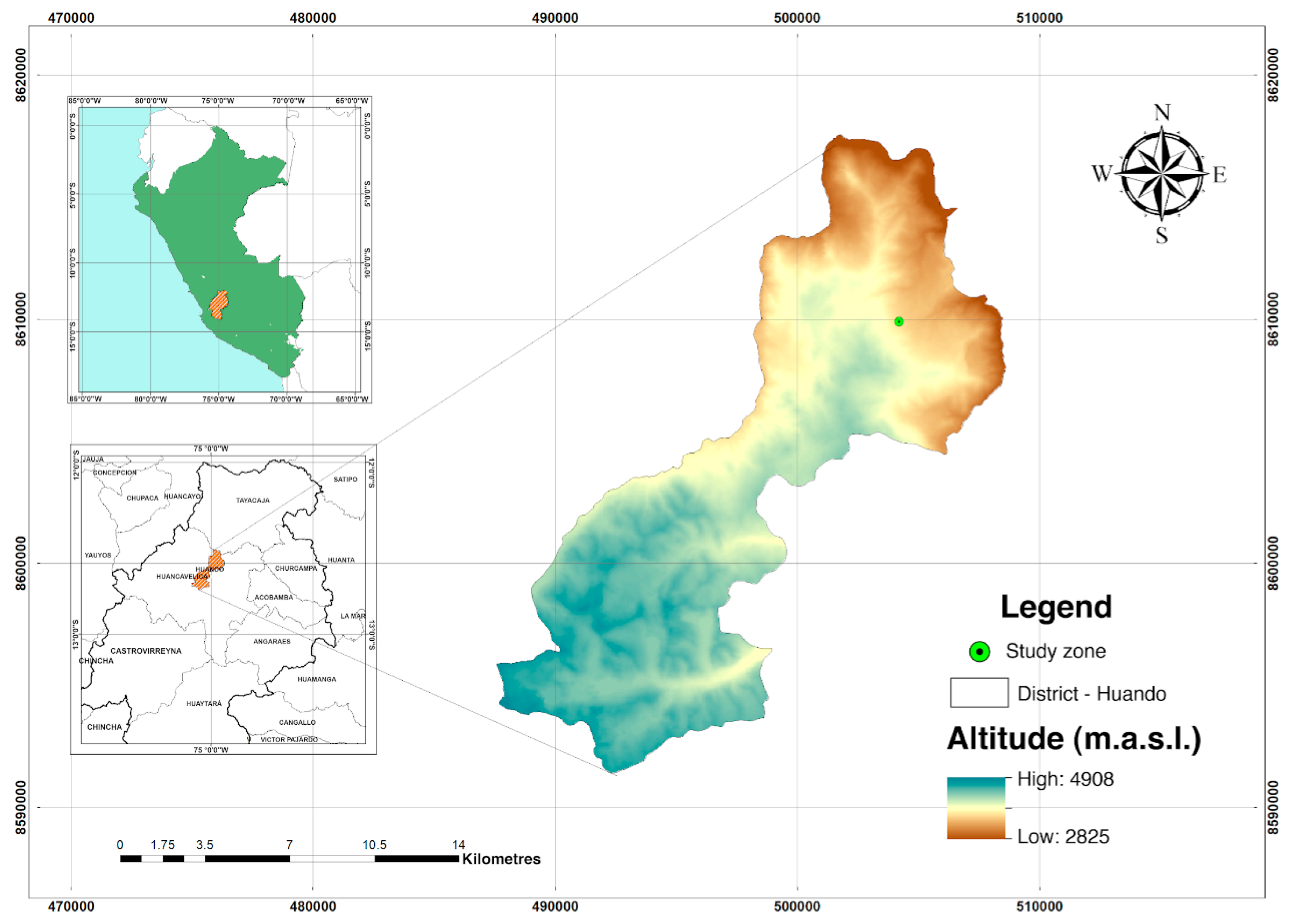
2.1. Field Site and Sampling
2.2. Soil Biogeochemical Properties
2.3. DNA Extraction and Illumina MiSeq Sequencing
2.4. Processing of 16S/ITS Sequences and Taxonomic Attribution
2.5. Statistics Analysis
3. Results
3.1. Sequencing Results and Quality Control
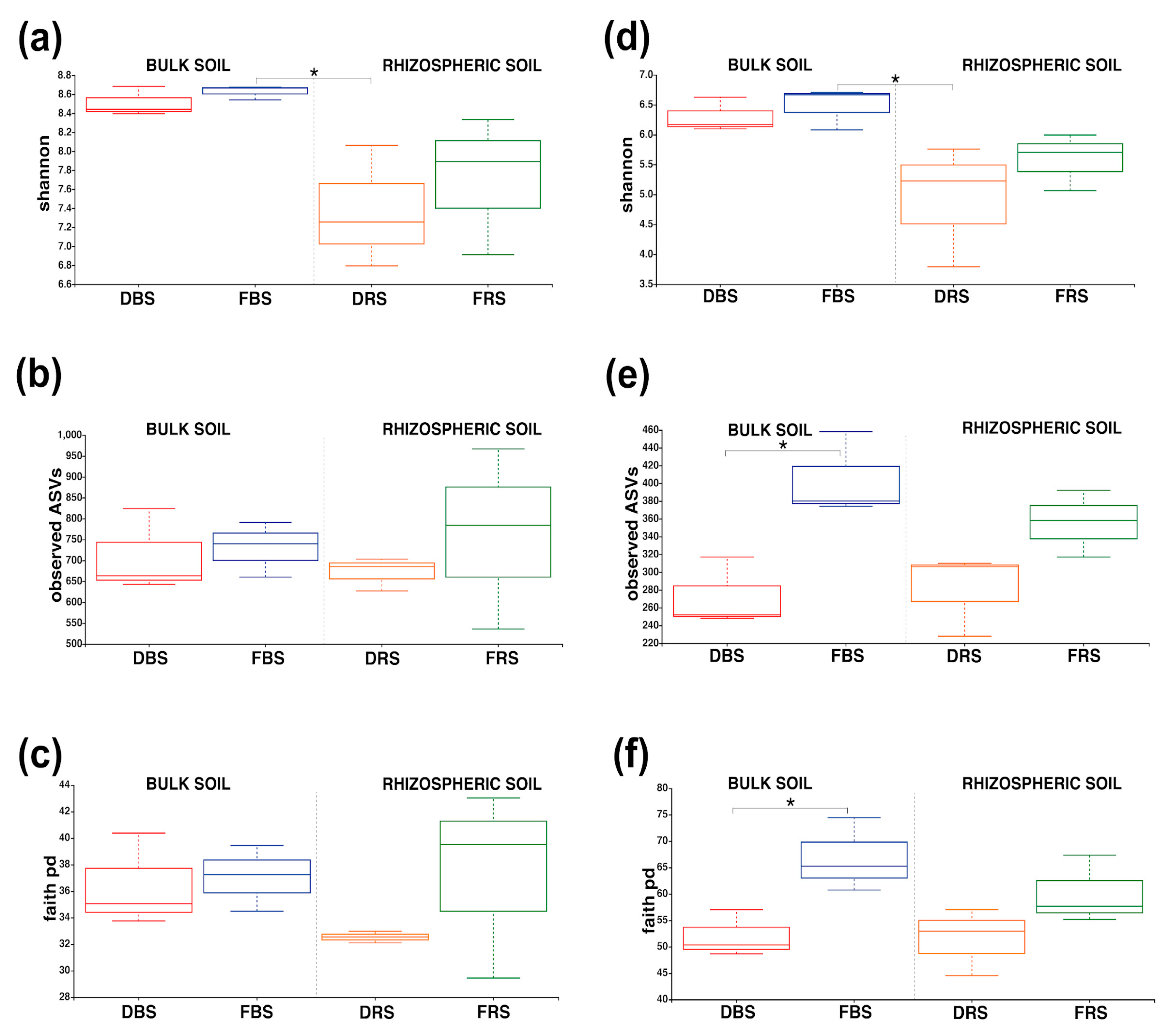
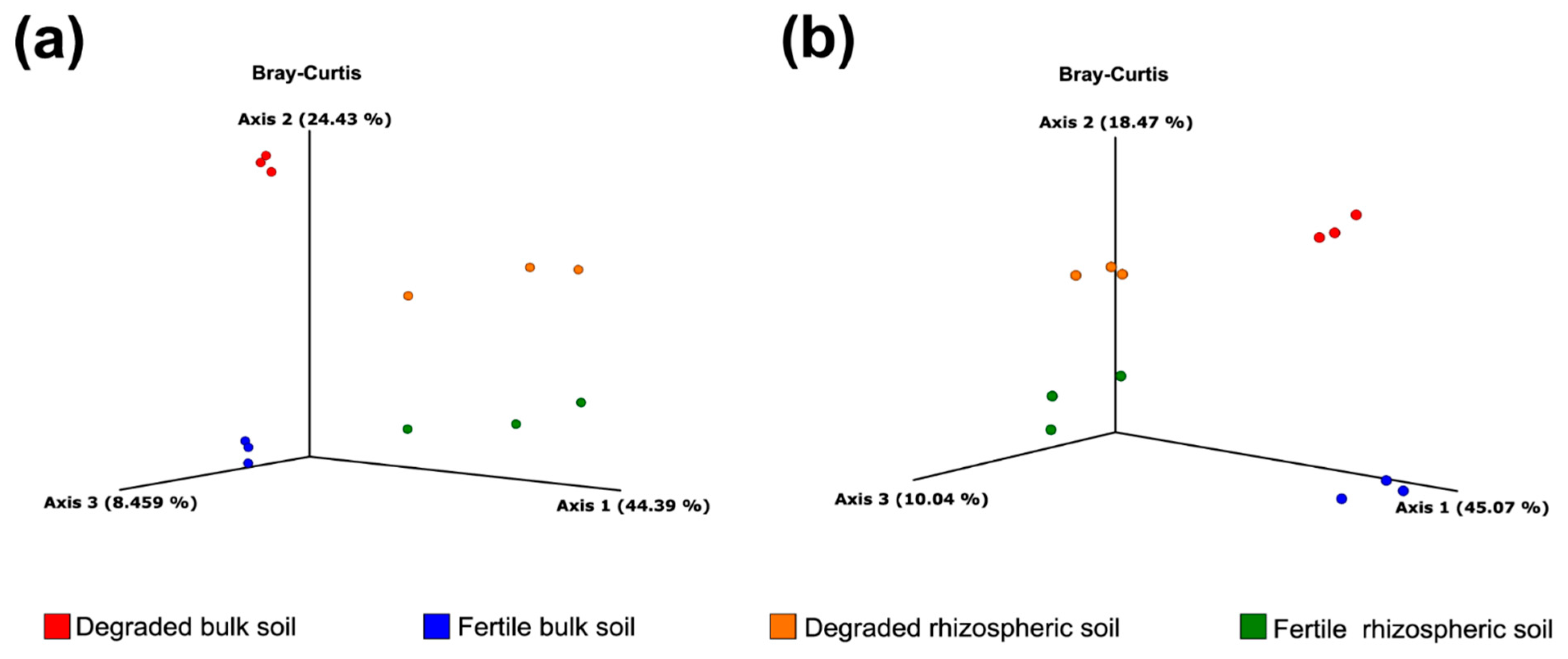
3.2. Alpha/Beta Diversity
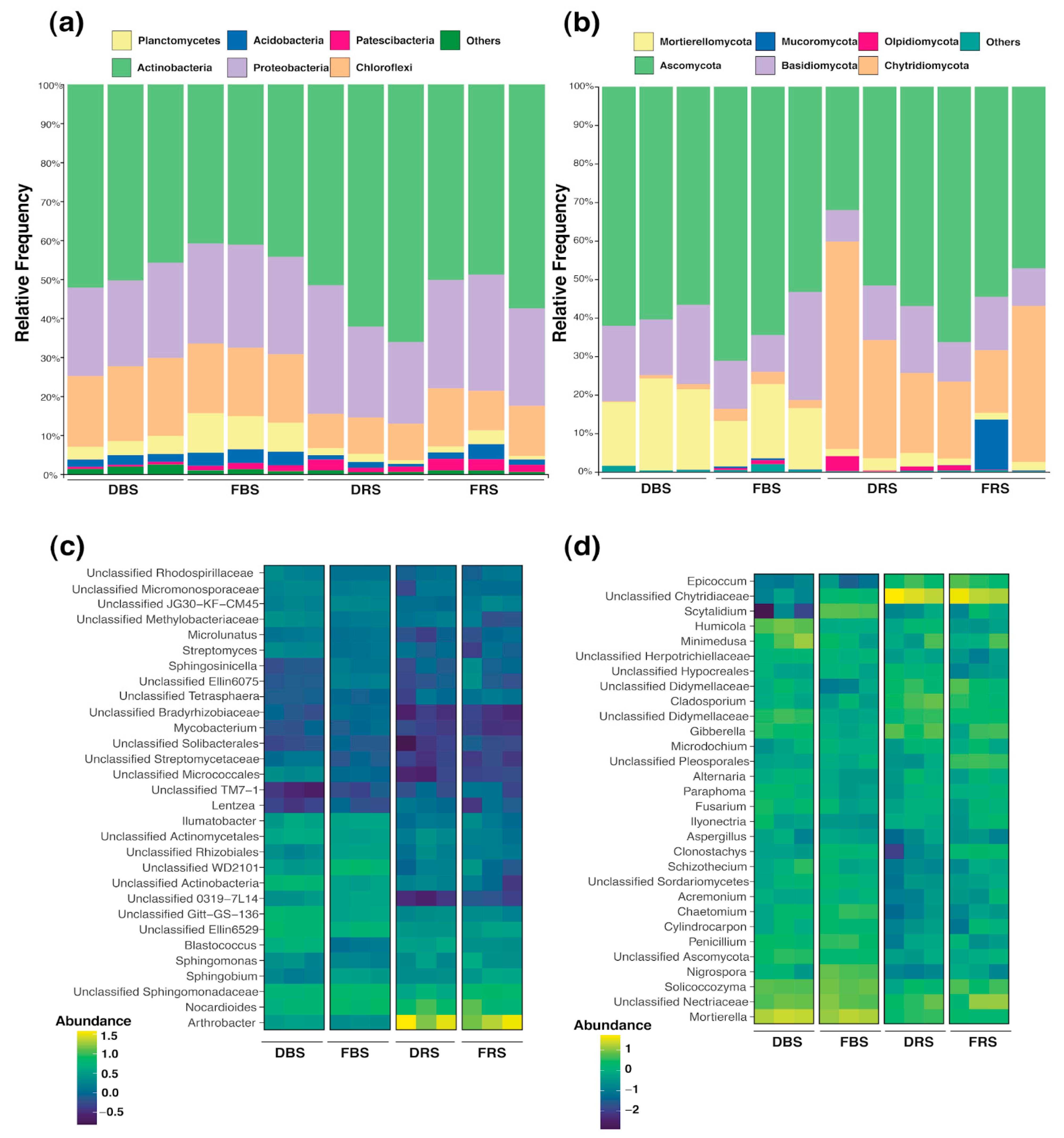
3.3. Taxonomic Composition of Fungal and Bacterial Communities in Rhizosphere Soil
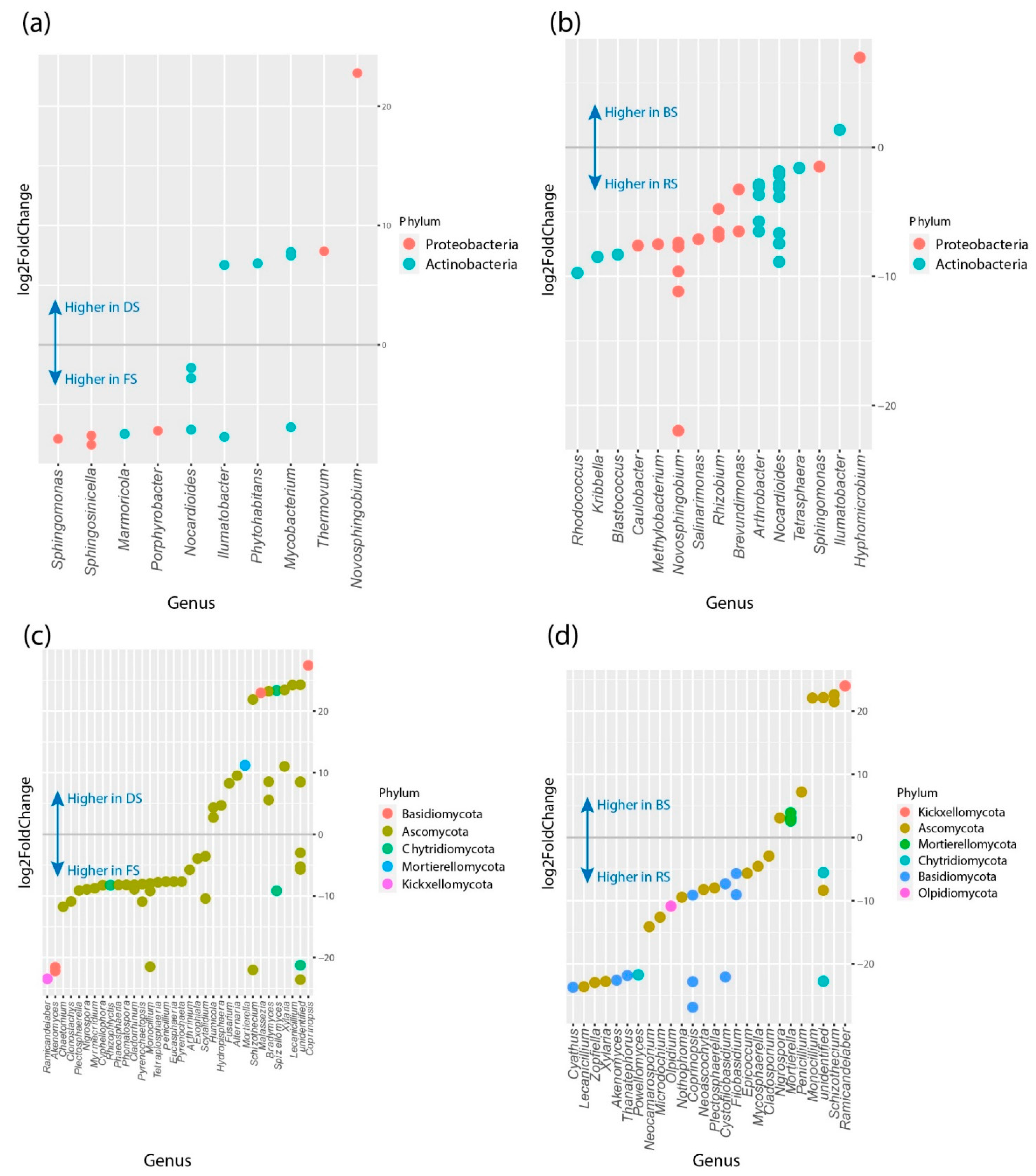
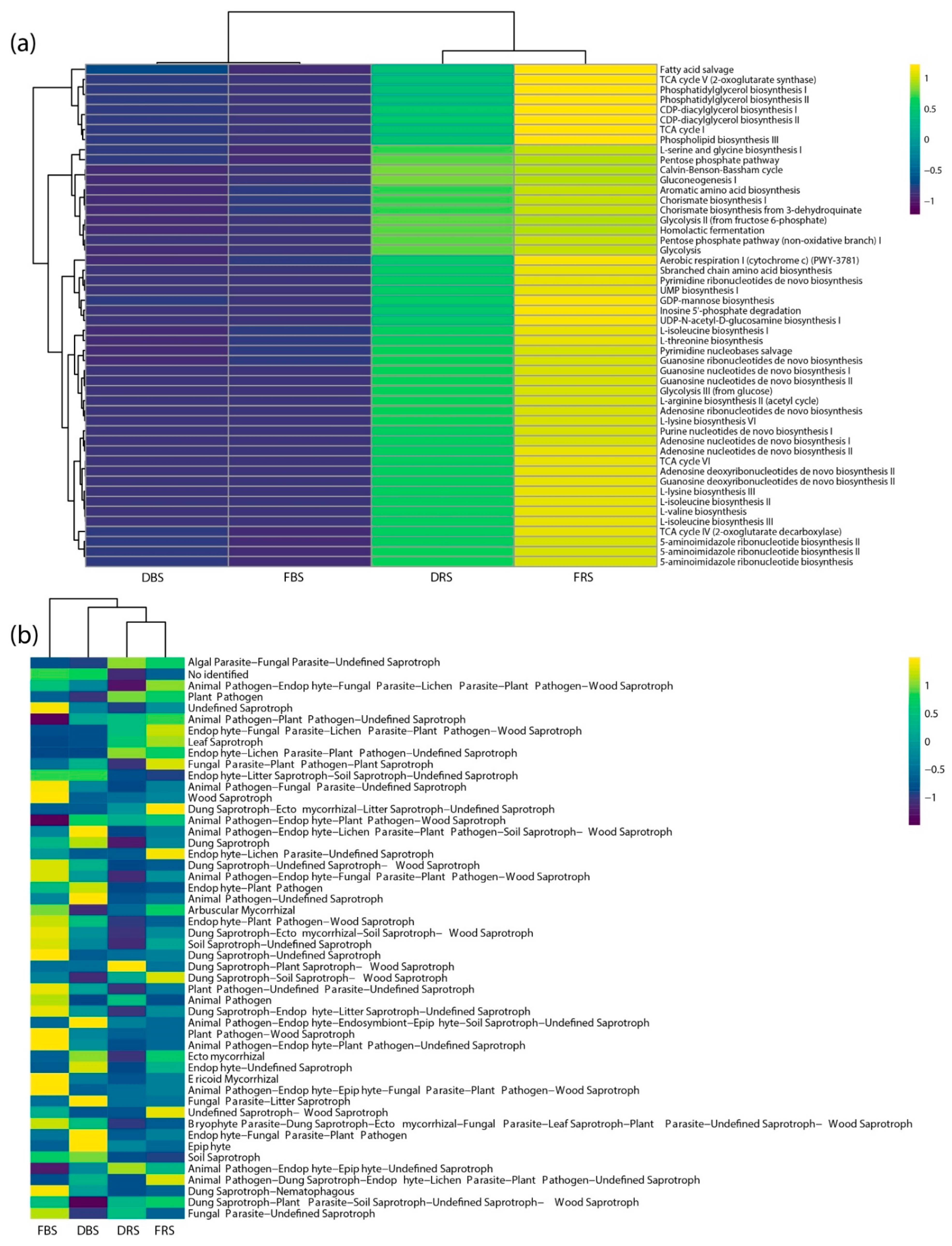
3.4. Differences in Relative Abundance and Functional Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemanceau, P.; Maron, P.-A.; Mazurier, S.; Mougel, C.; Pivato, B.; Plassart, P.; Ranjard, L.; Revellin, C.; Tardy, V.; Wipf, D. Understanding and Managing Soil Biodiversity: A Major Challenge in Agroecology. Agron. Sustain. Dev. 2015, 35, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Giller, K.E.; Beare, M.H.; Lavelle, P.; Izac, A.-M.N.; Swift, M.J. Agricultural Intensification, Soil Biodiversity and Agroecosystem Function. Appl. Soil Ecol. 1997, 6, 3–16. [Google Scholar] [CrossRef]
- Geisen, S.; Briones, M.J.I.; Gan, H.; Behan-Pelletier, V.M.; Friman, V.-P.; De Groot, G.A.; Hannula, S.E.; Lindo, Z.; Philippot, L.; Tiunov, A.V.; et al. A Methodological Framework to Embrace Soil Biodiversity. Soil Biol. Biochem. 2019, 136, 107536. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Reich, P.B.; Khachane, A.N.; Campbell, C.D.; Thomas, N.; Freitag, T.E.; Abu Al-Soud, W.; Sørensen, S.; Bardgett, R.D.; Singh, B.K. It Is Elemental: Soil Nutrient Stoichiometry Drives Bacterial Diversity: C:N:P Stoichiometry Drives Bacterial Diversity. Environ. Microbiol. 2017, 19, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolinska, A.; Frąc, M.; Oszust, K.; Szafranek-Nakonieczna, A.; Zielenkiewicz, U.; Stępniewska, Z. Microbial Biodiversity of Meadows under Different Modes of Land Use: Catabolic and Genetic Fingerprinting. World J. Microbiol. Biotechnol. 2017, 33, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Q.; Sui, J.; Zhang, J.; Liu, Z.; Du, J.; Xu, R.; Zhou, Y.; Liu, X. Isolation and Characterization of Antagonistic Bacteria Paenibacillus Jamilae HS-26 and Their Effects on Plant Growth. BioMed Res. Int. 2019, 2019, 3638926. [Google Scholar] [CrossRef] [Green Version]
- Nwachukwu, B.C.; Babalola, O.O. Metagenomics: A Tool for Exploring Key Microbiome With the Potentials for Improving Sustainable Agriculture. Front. Sustain. Food Syst. 2022, 6, 886987. [Google Scholar] [CrossRef]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of Diet to the Composition of the Human Gut Microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef]
- Pathan, S.; Siddiqui, R.A. Nutritional Composition and Bioactive Components in Quinoa (Chenopodium Quinoa Willd.) Greens: A Review. Nutrients 2022, 14, 558. [Google Scholar] [CrossRef]
- Bedoya-Perales, N.S.; Pumi, G.; Mujica, A.; Talamini, E.; Padula, A.D. Quinoa Expansion in Peru and Its Implications for Land Use Management. Sustainability 2018, 10, 532. [Google Scholar] [CrossRef] [Green Version]
- Alandia, G.; Rodriguez, J.P.; Jacobsen, S.-E.; Bazile, D.; Condori, B. Global Expansion of Quinoa and Challenges for the Andean Region. Glob. Food Secur. 2020, 26, 100429. [Google Scholar] [CrossRef]
- Phillips, R.P.; Fahey, T.J. Tree Species and Mycorrhizal Associations Influence the Magnitude of Rhizosphere Effects. Ecology 2006, 87, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- García-Mantrana, I.; Selma-Royo, M.; González, S.; Parra-Llorca, A.; Martínez-Costa, C.; Collado, M.C. Distinct Maternal Microbiota Clusters Are Associated with Diet during Pregnancy: Impact on Neonatal Microbiota and Infant Growth during the First 18 Months of Life. Gut Microbes 2020, 11, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Mesonero, L.; Ferrús, M.A.; Moreno, Y. Determination of the Bacterial Microbiome of Free-Living Amoebae Isolated from Wastewater by 16S RRNA Amplicon-Based Sequencing. Environ. Res. 2020, 190, 109987. [Google Scholar] [CrossRef]
- Asemaninejad, A.; Weerasuriya, N.; Gloor, G.B.; Lindo, Z.; Thorn, R.G. New Primers for Discovering Fungal Diversity Using Nuclear Large Ribosomal DNA. PLoS ONE 2016, 11, e0159043. [Google Scholar] [CrossRef] [Green Version]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-Coverage ITS Primers for the DNA-Based Identification of Ascomycetes and Basidiomycetes in Environmental Samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Hui, H.; Shao, T.; Gao, X.; Long, X.; Rengel, Z. Effects of Planting Quinoa on Soil Properties and Microbial Community in Saline Soil. Authorea 2022, preprints. [Google Scholar] [CrossRef]
- Raiger Iustman, L.J.; Almasqué, F.J.; Vullo, D.L. Microbiota Diversity Change as Quality Indicator of Soils Exposed to Intensive Periurban Agriculture. Curr. Microbiol. 2021, 78, 338–346. [Google Scholar] [CrossRef]
- Zhao, Q.; Xiong, W.; Xing, Y.; Sun, Y.; Lin, X.; Dong, Y. Long-Term Coffee Monoculture Alters Soil Chemical Properties and Microbial Communities. Sci. Rep. 2018, 8, 6116. [Google Scholar] [CrossRef] [Green Version]
- Pertile, G.; Panek, J.; Oszust, K.; Siczek, A.; Oleszek, M.; Gryta, A.; Frąc, M. Effect of Different Organic Waste on Cellulose-Degrading Enzymes Secreted by Petriella Setifera in the Presence of Cellobiose and Glucose. Cellulose 2019, 26, 7905–7922. [Google Scholar] [CrossRef] [Green Version]
- Zeiner, C.A.; Purvine, S.O.; Zink, E.M.; Paša-Tolić, L.; Chaput, D.L.; Haridas, S.; Wu, S.; LaButti, K.; Grigoriev, I.V.; Henrissat, B.; et al. Comparative Analysis of Secretome Profiles of Manganese(II)-Oxidizing Ascomycete Fungi. PLoS ONE 2016, 11, e0157844. [Google Scholar] [CrossRef] [Green Version]
- Song, A.; Zhang, J.; Xu, D.; Wang, E.; Bi, J.; Asante-Badu, B.; Njyenawe, M.C.; Sun, M.; Xue, P.; Wang, S.; et al. Keystone Microbial Taxa Drive the Accelerated Decompositions of Cellulose and Lignin by Long-Term Resource Enrichments. Sci. Total Environ. 2022, 842, 156814. [Google Scholar] [CrossRef]
- Negash, M.; Kaseva, J.; Kahiluoto, H. Perennial Monocropping of Khat Decreased Soil Carbon and Nitrogen Relative to Multistrata Agroforestry and Natural Forest in Southeastern Ethiopia. Reg. Environ. Chang. 2022, 22, 38. [Google Scholar] [CrossRef]
- Rosset, J.S.; Lana, M.d.C.; Pereira, M.G.; Schiavo, J.A.; Rampim, L.; Sarto, M.V.M. Frações químicas e oxidáveis da matéria orgânica do solo sob diferentes sistemas de manejo, em Latossolo Vermelho. Pesqui. Agropecuária Bras. 2016, 51, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Loss, A.; Pereira, M.G.; Perin, A.; Beutler, S.J.; Anjos, L.H.C. dos Oxidizable Carbon and Humic Substances in Rotation Systems with Brachiaria/Livestock and Pearl Millet/No Livestock in the Brazilian Cerrado. Span. J. Agric. Res. 2013, 11, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hou, R.; Gong, Y.; Li, H.; Fan, M.; Kuzyakov, Y. Effects of 11 Years of Conservation Tillage on Soil Organic Matter Fractions in Wheat Monoculture in Loess Plateau of China. Soil Tillage Res. 2009, 106, 85–94. [Google Scholar] [CrossRef]
- Strous, M.; Fuerst, J.A.; Kramer, E.H.M.; Logemann, S.; Muyzer, G.; van de Pas-Schoonen, K.T.; Webb, R.; Kuenen, J.G.; Jetten, M.S.M. Missing Lithotroph Identified as New Planctomycete. Nature 1999, 400, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Pizzetti, I.; Fuchs, B.M.; Gerdts, G.; Wichels, A.; Wiltshire, K.H.; Amann, R. Temporal Variability of Coastal Planctomycetes Clades at Kabeltonne Station, North Sea. Appl. Environ. Microbiol. 2011, 77, 5009–5017. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Pang, Z.; Fallah, N.; Zhou, Y.; Zhang, C.; Hu, C.; Lin, W.; Yuan, Z. Diversity of Microbial Communities and Soil Nutrients in Sugarcane Rhizosphere Soil under Water Soluble Fertilizer. PLoS ONE 2021, 16, e0245626. [Google Scholar] [CrossRef]
- Tan, Y.; Cui, Y.; Li, H.; Kuang, A.; Li, X.; Wei, Y.; Ji, X. Rhizospheric Soil and Root Endogenous Fungal Diversity and Composition in Response to Continuous Panax Notoginseng Cropping Practices. Microbiol. Res. 2017, 194, 10–19. [Google Scholar] [CrossRef]
- Chen, M.; Li, X.; Yang, Q.; Chi, X.; Pan, L.; Chen, N.; Yang, Z.; Wang, T.; Wang, M.; Yu, S. Soil Eukaryotic Microorganism Succession as Affected by Continuous Cropping of Peanut—Pathogenic and Beneficial Fungi Were Selected. PLoS ONE 2012, 7, e40659. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Zhao, Q.; Zhao, J.; Xun, W.; Li, R.; Zhang, R.; Wu, H.; Shen, Q. Different Continuous Cropping Spans Significantly Affect Microbial Community Membership and Structure in a Vanilla-Grown Soil as Revealed by Deep Pyrosequencing. Microb. Ecol. 2015, 70, 209–218. [Google Scholar] [CrossRef]
- Sergaki, C.; Lagunas, B.; Lidbury, I.; Gifford, M.L.; Schäfer, P. Challenges and Approaches in Microbiome Research: From Fundamental to Applied. Front. Plant Sci. 2018, 9, 1205. [Google Scholar] [CrossRef]
- Onwona-Kwakye, M.; Plants-Paris, K.; Keita, K.; Lee, J.; Brink, P.J.V.D.; Hogarh, J.N.; Darkoh, C. Pesticides Decrease Bacterial Diversity and Abundance of Irrigated Rice Fields. Microorganisms 2020, 8, 318. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Fu, H.; Zhu, B.; Hussain, H.A.; Zhang, K.; Tian, X.; Duan, M.; Xie, X.; Wang, L. Potassium Improves Drought Stress Tolerance in Plants by Affecting Root Morphology, Root Exudates and Microbial Diversity. Metabolites 2021, 11, 131. [Google Scholar] [CrossRef]
- Landa, M.; Cottrell, M.T.; Kirchman, D.L.; Blain, S.; Obernosterer, I. Changes in Bacterial Diversity in Response to Dissolved Organic Matter Supply in a Continuous Culture Experiment. Aquat. Microb. Ecol. 2013, 69, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Sinsabaugh, R.L.; Foreman, C.M. 18—Integrating Dissolved Organic Matter Metabolism and Microbial Diversity: An Overview of Conceptual Models. In Aquatic Ecosystems; Findlay, S.E.G., Sinsabaugh, R.L., Eds.; Aquatic Ecology; Academic Press: Burlington, NJ, USA, 2003; pp. 425–454. ISBN 978-0-12-256371-3. [Google Scholar]
- Lee, Y.-H.; Ahn, B.-K.; Sonn, Y.-K. Effects of Electrical Conductivity on the Soil Microbial Community in a Controled Horticultural Land for Strawberry Cultivation. Korean J. Soil Sci. Fertil. 2011, 44, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, L.; Ling, N.; Zhu, C.; Chi, F.; Li, W.; Hao, X.; Zhang, W.; Bian, J.; Chen, L.; et al. Exploring Soil Factors Determining Composition and Structure of the Bacterial Communities in Saline-Alkali Soils of Songnen Plain. Front. Microbiol. 2019, 10, 2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities Along an Altitudinal Gradient in Alpine Forest Soils: What Are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Fu, W.; Hu, C.; Chen, G.; Xiao, Z.; Chen, Y.; Wang, Z.; Cheng, H. Variation of Rhizosphere Microbial Community in Continuous Mono-Maize Seed Production. Sci. Rep. 2021, 11, 1544. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, U.B.; Sahu, P.K.; Paul, S.; Kumar, A.; Malviya, D.; Singh, S.; Kuppusamy, P.; Singh, P.; Paul, D.; et al. Linking Soil Microbial Diversity to Modern Agriculture Practices: A Review. Int. J. Environ. Res. Public. Health 2022, 19, 3141. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, N.; Lata, P.; Dwivedi, V.; Singh, A.; Niharika, N.; Kaur, J.; Anand, S.; Malhotra, J.; Jindal, S.; Nigam, A.; et al. Comparative Metagenomic Analysis of Soil Microbial Communities across Three Hexachlorocyclohexane Contamination Levels. PLoS ONE 2012, 7, e46219. [Google Scholar] [CrossRef]
- Okazaki, K.; Tsurumaru, H.; Hashimoto, M.; Takahashi, H.; Okubo, T.; Ohwada, T.; Minamisawa, K.; Ikeda, S. Community Analysis-Based Screening of Plant Growth-Promoting Bacteria for Sugar Beet. Microbes Environ. 2021, 36, ME20137. [Google Scholar] [CrossRef]
- Zytynska, S.E.; Eicher, M.; Rothballer, M.; Weisser, W.W. Microbial-Mediated Plant Growth Promotion and Pest Suppression Varies Under Climate Change. Front. Plant Sci. 2020, 11, 573578. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Verma, J.P. The Role of Microbes to Improve Crop Productivity and Soil Health. In Ecological Wisdom Inspired Restoration Engineering; Achal, V., Mukherjee, A., Eds.; EcoWISE; Springer: Singapore, 2019; pp. 249–265. ISBN 9789811301483. [Google Scholar]
- Hu, J.; Wei, Z.; Kowalchuk, G.A.; Xu, Y.; Shen, Q.; Jousset, A. Rhizosphere Microbiome Functional Diversity and Pathogen Invasion Resistance Build up during Plant Development. Environ. Microbiol. 2020, 22, 5005–5018. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ding, C.; Zhang, T.; Wang, X. Fungal Pathogen Accumulation at the Expense of Plant-Beneficial Fungi as a Consequence of Consecutive Peanut Monoculturing. Soil Biol. Biochem. 2014, 72, 11–18. [Google Scholar] [CrossRef]
- Adnan, M.; Islam, W.; Gang, L.; Chen, H.Y.H. Advanced Research Tools for Fungal Diversity and Its Impact on Forest Ecosystem. Environ. Sci. Pollut. Res. 2022, 29, 45044–45062. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | R2 | F | Pr (>F) | |
|---|---|---|---|---|
| Bacterial | Kind of soil | 0.3372 | 7.3989 | 0.001 * |
| Monocropping | 0.1659 | 3.6412 | 0.035 * | |
| Kind of soil × Monocropping | 0.1323 | 2.9050 | 0.048 * | |
| Fungal | Kind of soil | 0.4111 | 9.3141 | 0.001 * |
| Monocropping | 0.1618 | 3.6651 | 0.011 * | |
| Kind of soil × Monocropping | 0.0740 | 1.6768 | 0.107 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada, R.; Cosme, R.; Porras, T.; Reynoso, A.; Calderon, C.; Arbizu, C.I.; Arone, G.J. Changes in Bulk and Rhizosphere Soil Microbial Diversity Communities of Native Quinoa Due to the Monocropping in the Peruvian Central Andes. Microorganisms 2023, 11, 1926. https://doi.org/10.3390/microorganisms11081926
Estrada R, Cosme R, Porras T, Reynoso A, Calderon C, Arbizu CI, Arone GJ. Changes in Bulk and Rhizosphere Soil Microbial Diversity Communities of Native Quinoa Due to the Monocropping in the Peruvian Central Andes. Microorganisms. 2023; 11(8):1926. https://doi.org/10.3390/microorganisms11081926
Chicago/Turabian StyleEstrada, Richard, Roberto Cosme, Tatiana Porras, Auristela Reynoso, Constatino Calderon, Carlos I. Arbizu, and Gregorio J. Arone. 2023. "Changes in Bulk and Rhizosphere Soil Microbial Diversity Communities of Native Quinoa Due to the Monocropping in the Peruvian Central Andes" Microorganisms 11, no. 8: 1926. https://doi.org/10.3390/microorganisms11081926
APA StyleEstrada, R., Cosme, R., Porras, T., Reynoso, A., Calderon, C., Arbizu, C. I., & Arone, G. J. (2023). Changes in Bulk and Rhizosphere Soil Microbial Diversity Communities of Native Quinoa Due to the Monocropping in the Peruvian Central Andes. Microorganisms, 11(8), 1926. https://doi.org/10.3390/microorganisms11081926