

Whole Genome Sequencing and Pan-Genomic Analysis of Multidrug-Resistant Vibrio cholerae VC01 Isolated from a Clinical Sample

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Antibiotics Susceptibility
2.2. Extraction of Genomic DNA and Whole Genome Sequencing
2.3. Type Strain-Based Identification, MLST, and Whole Genome Annotation
2.4. Detection of Mobile Genetic Elements
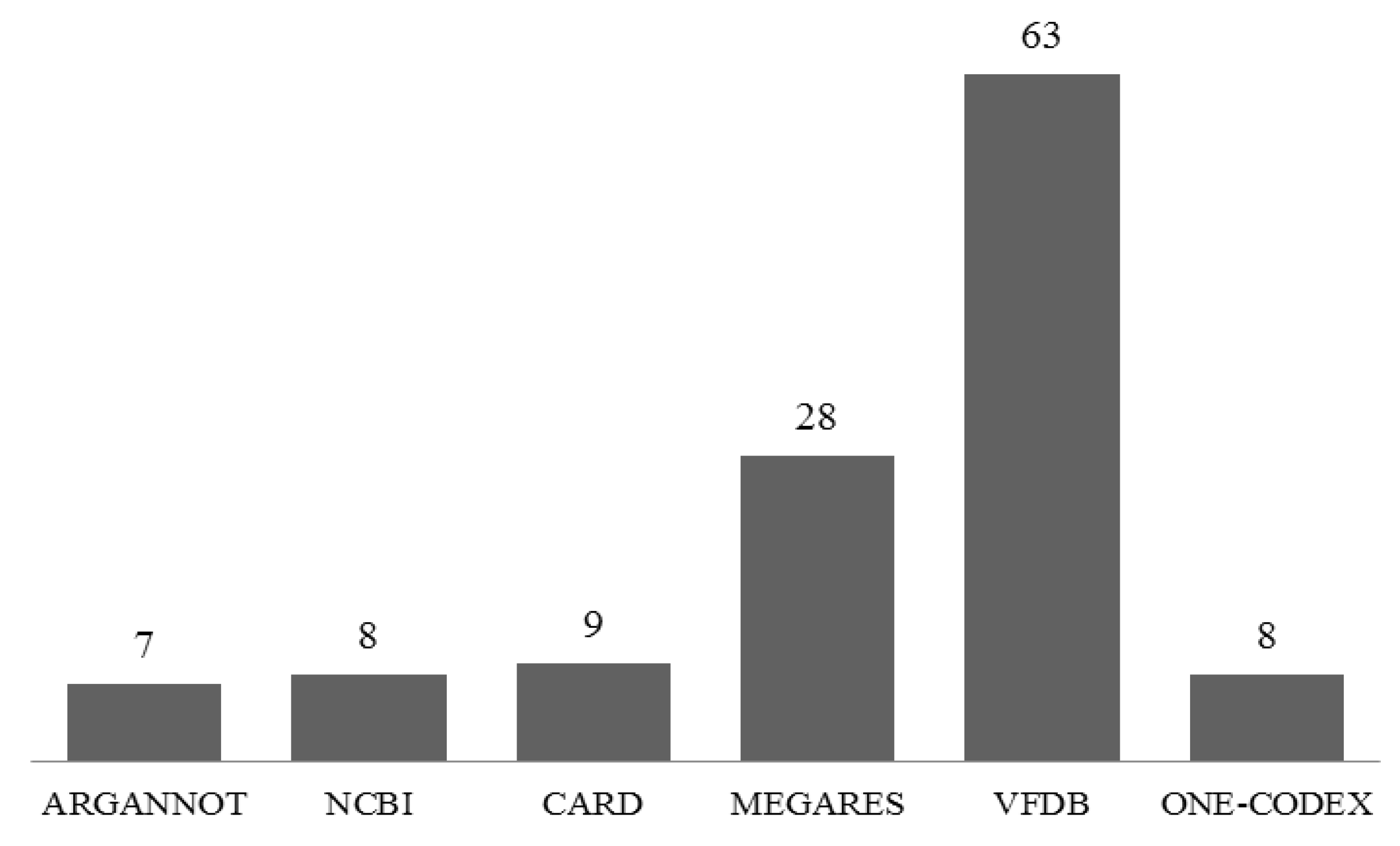
2.5. Identification of the Antimicrobial Resistance (AMR) Gene
2.6. Pan-Genome Analysis
3. Results
3.1. Isolation and Antimicrobial Susceptibility
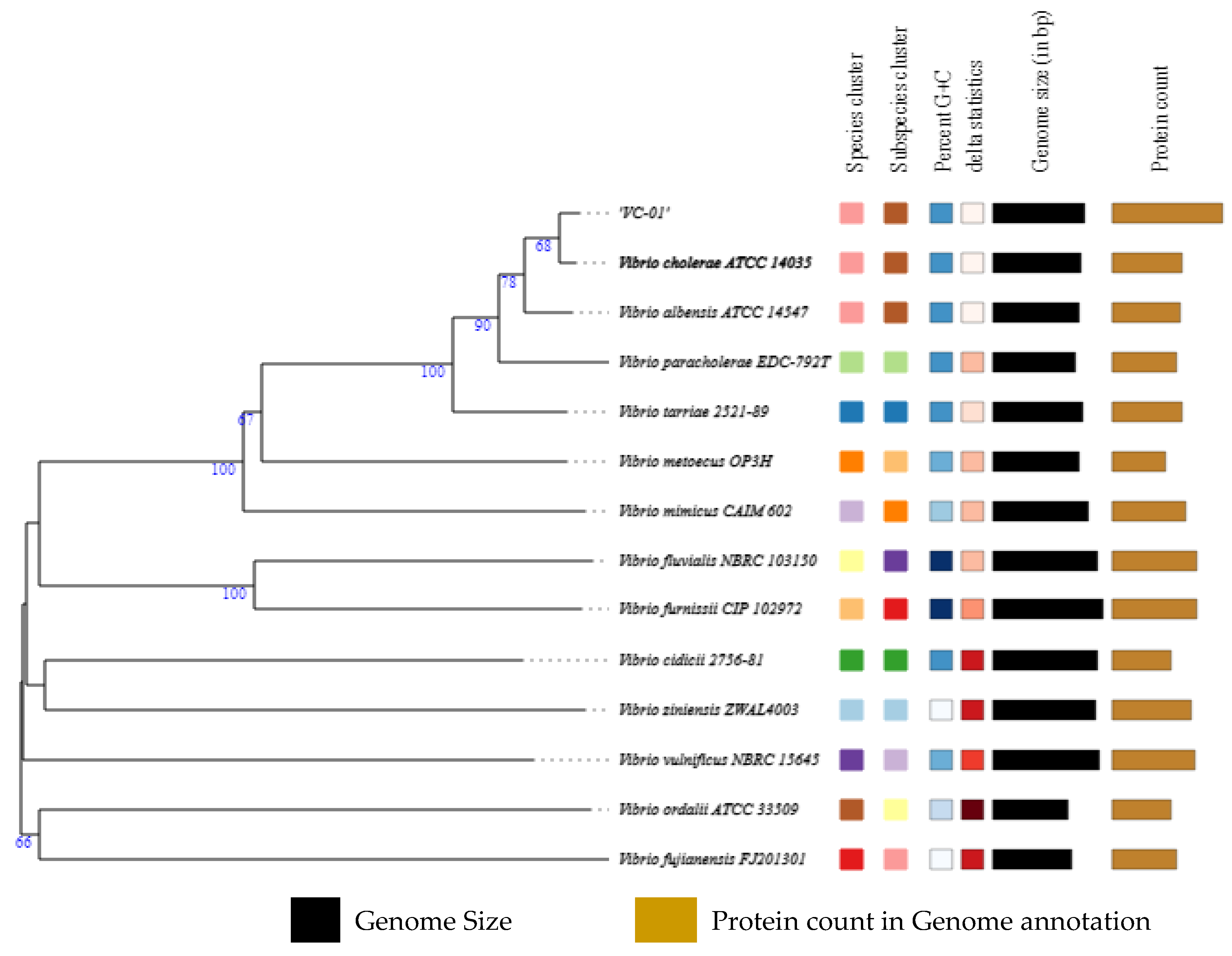
3.2. Genome Assembly and Identification
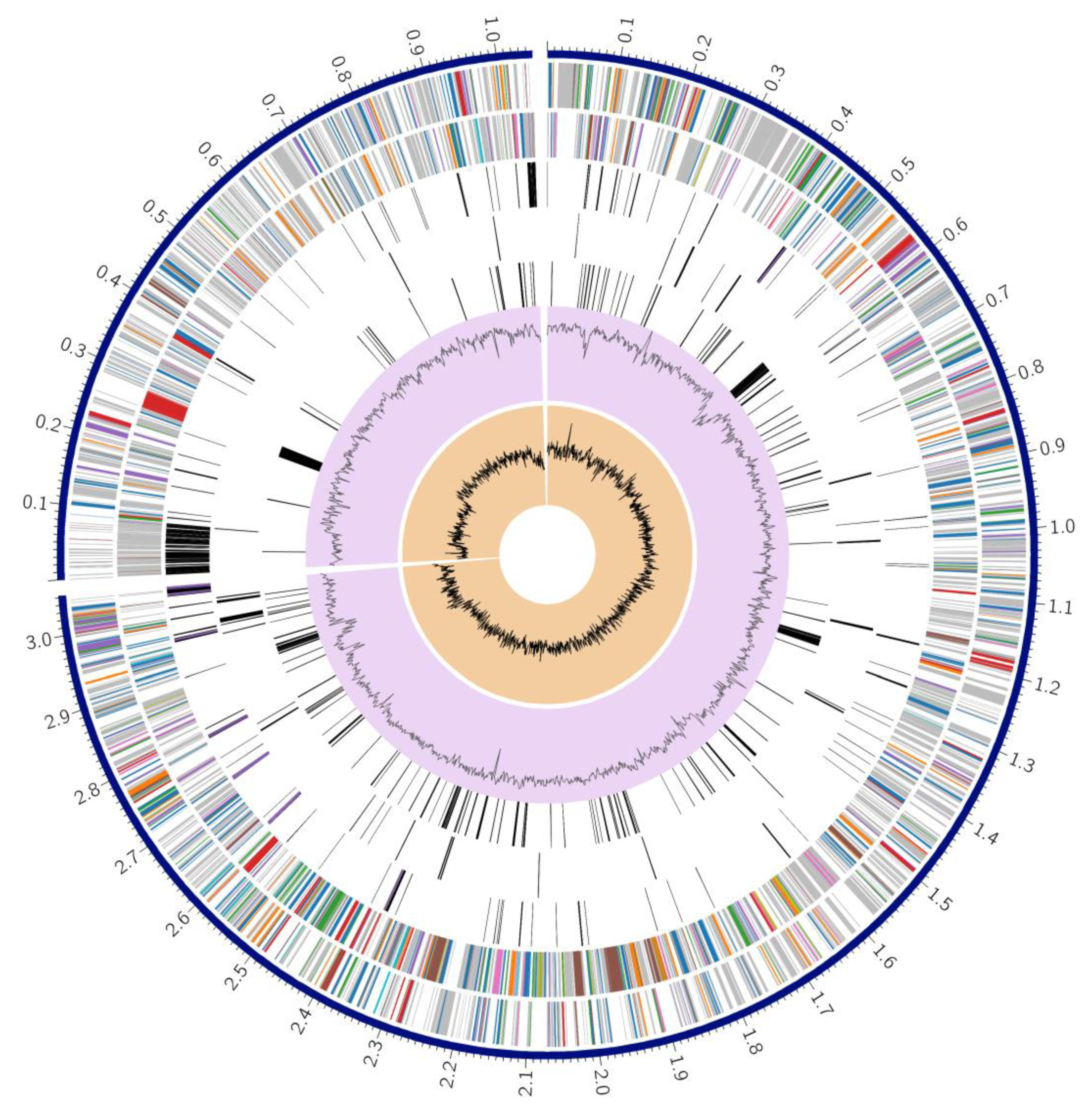
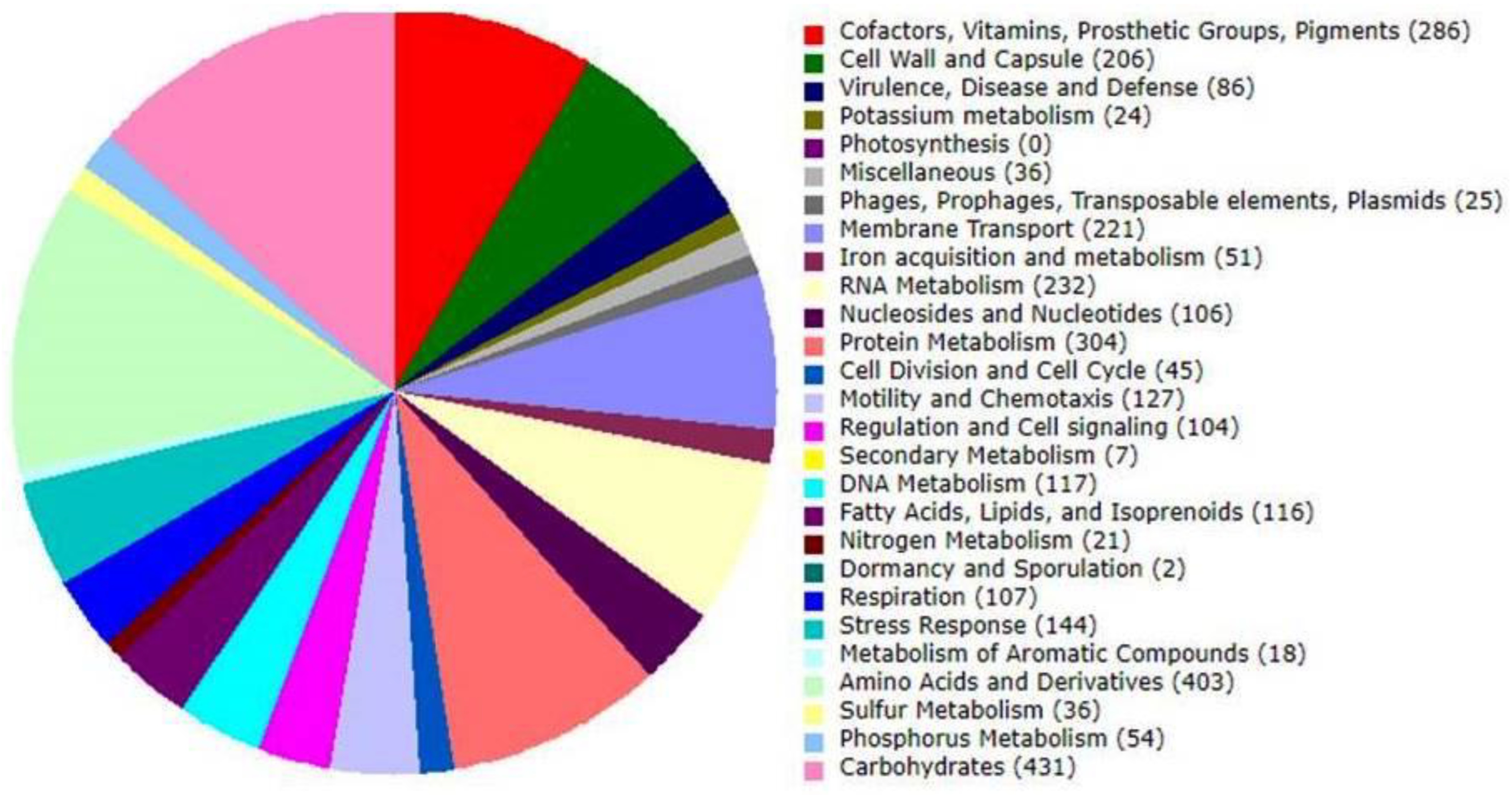
3.3. Genome Annotation
3.4. Identification of Antibiotic Resistance Genes
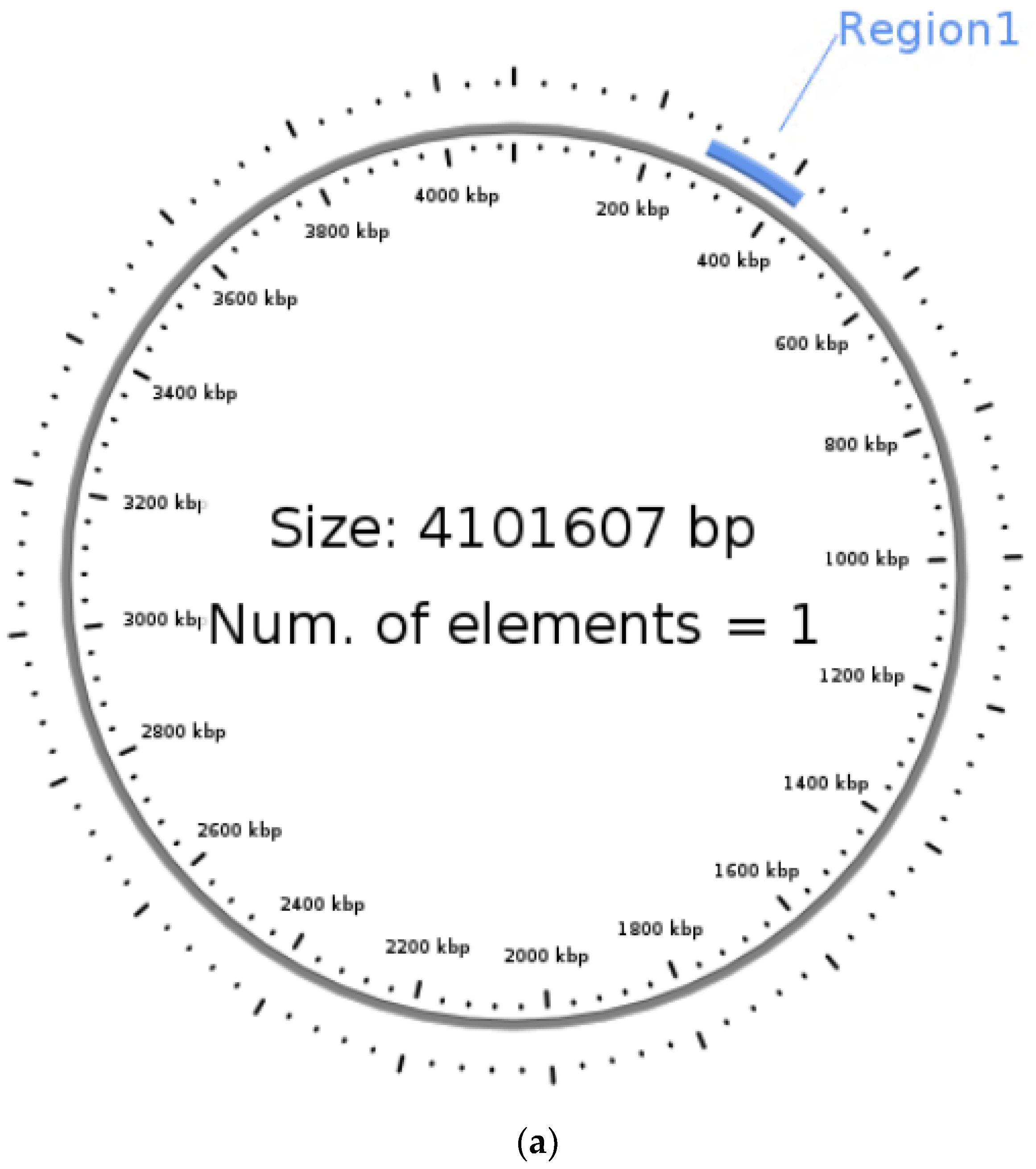
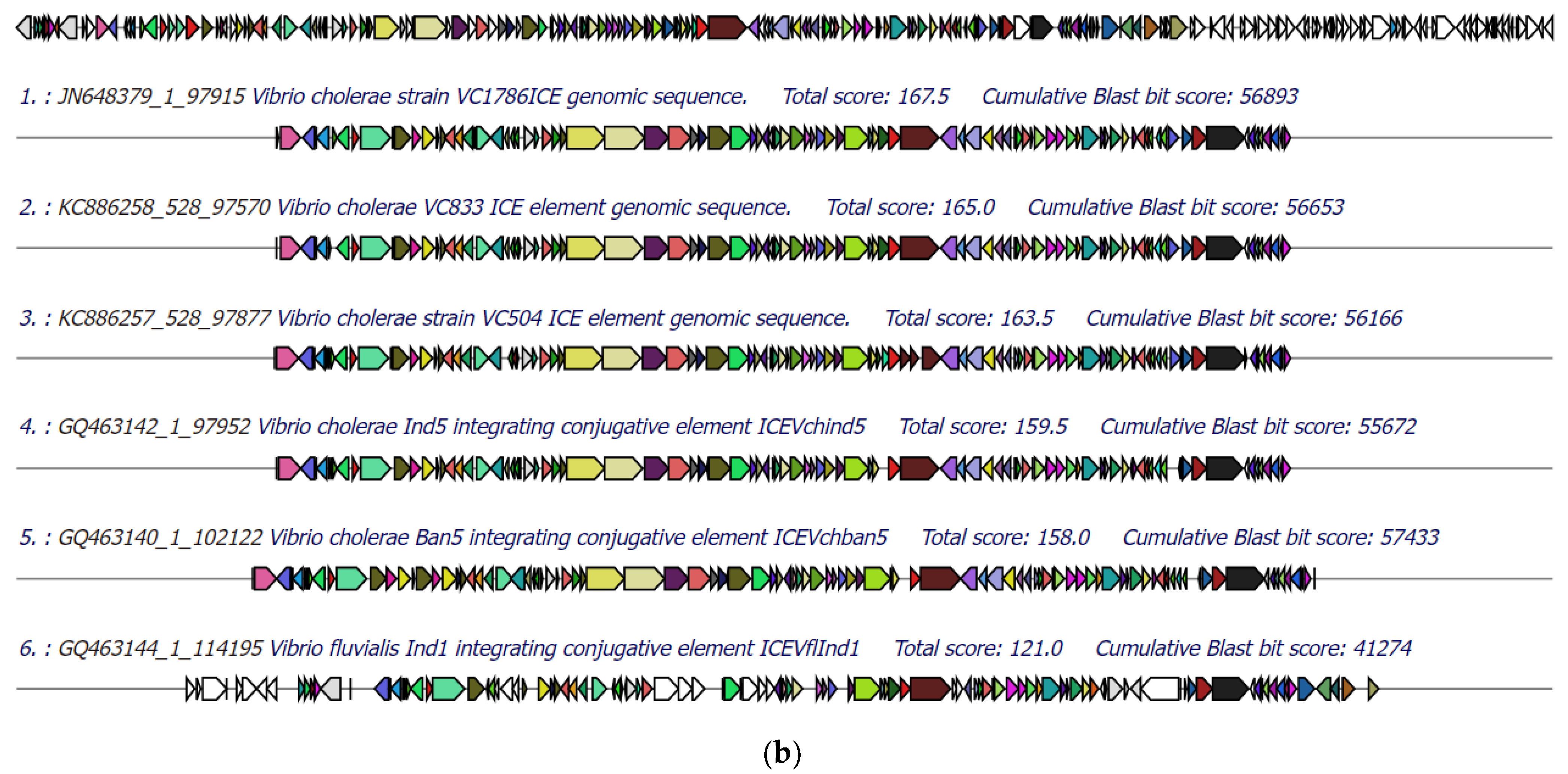
3.5. Identification of Integrative Conjugative Element (ICE)
3.6. Identification and Comparison of Prophage Sequences
3.7. Pan-Core Genome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, M.; Nelson, A.R.; Lopez, A.L.; Sack, D.A. Updated Global Burden of Cholera in Endemic Countries. PLoS Negl. Trop. Dis. 2015, 9, e0003832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Liu, B.; Feng, L.; Ding, P.; Guo, X.; Wang, M.; Cao, B.; Reeves, P.R.; Wang, L. Origins of the Current Seventh Cholera Pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, E7730–E7739. [Google Scholar] [CrossRef] [PubMed]
- Baranova, D.E.; Levinson, K.J.; Mantis, N.J. Vibrio Cholerae O1 Secretes an Extracellular Matrix in Response to Antibody-Mediated Agglutination. PLoS ONE 2018, 13, e0190026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Zhu, L.-W.W.; Jing, J.; Guan, J.Y.; Lu, G.-J.J.; Xie, L.-H.H.; Ji, X.; Chu, D.; Sun, Y.; Chen, P.; et al. Pan-Genome Analysis of Vibrio Cholerae and Vibrio Metschnikovii Strains Isolated From Migratory Birds at Dali Nouer Lake in Chifeng, China. Front. Vet. Sci. 2021, 8, 638820. [Google Scholar] [CrossRef]
- Devault, A.M.; Golding, G.B.; Waglechner, N.; Enk, J.M.; Kuch, M.; Tien, J.H.; Shi, M.; Fisman, D.N.; Dhody, A.N.; Forrest, S.; et al. Second-Pandemic Strain of Vibrio Cholerae from the Philadelphia Cholera Outbreak of 1849. N. Engl. J. Med. 2014, 370, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, M.K.; Bhattacharya, S.K.; Garg, S.; Saha, P.K.; Dutta, D.; Balakrish Nair, G.; Deb, B.C.; Das, K.P.; Shimada, T.; Balakrish Nair, G.; et al. Outbreak of Vibrio Cholerae Non-01 in India and Bangladesh. Lancet 1993, 341, 1346–1347. [Google Scholar] [CrossRef]
- Faizi, N.; Kazmi, S. Universal Health Coverage—There Is More to It than Meets the Eye. J. Fam. Med. Prim. Care 2017, 6, 169–170. [Google Scholar] [CrossRef]
- Ferdous, J.; Sultana, R.; Rashid, R.B.; Tasnimuzzaman, M.; Nordland, A.; Begum, A.; Jensen, P.K.M. A Comparative Analysis of Vibrio Cholerae Contamination in Point-of-Drinking and Source Water in a Low-Income Urban Community, Bangladesh. Front. Microbiol. 2018, 9, 489. [Google Scholar] [CrossRef] [Green Version]
- Syngkon, A.; Elluri, S.; Koley, H.; Rompikuntal, P.K.; Saha, D.R.; Chakrabarti, M.K.; Bhadra, R.K.; Wai, S.N.; Pal, A. Studies on a Novel Serine Protease of a ΔhapaΔprtV Vibrio Cholerae O1 Strain and Its Role in Hemorrhagic Response in the Rabbit Ileal Loop Model. PLoS ONE 2010, 5, e13122. [Google Scholar] [CrossRef] [Green Version]
- Rajpara, N.; Vinothkumar, K.; Mohanty, P.; Singh, A.K.; Singh, R.; Sinha, R.; Nag, D.; Koley, H.; Kushwaha Bhardwaj, A. Synergistic Effect of Various Virulence Factors Leading to High Toxicity of Environmental V. Cholerae Non-O1/ Non-O139 Isolates Lacking Ctx Gene: Comparative Study with Clinical Strains. PLoS ONE 2013, 8, e76200. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, E.; Ochi, S.; Mizuno, T.; Morita, D.; Morita, M.; Ohnishi, M.; Koley, H.; Dutta, M.; Chowdhury, G.; Mukhopadhyay, A.K.; et al. Virulence of Cholera Toxin Gene-Positive Vibrio Cholerae Non-O1/Non-O139 Strains Isolated From Environmental Water in Kolkata, India. Front. Microbiol. 2021, 12, 726273. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Recommendations for the Use of Antibiotics for the Treatment of Cholera. 2018. Available online: https://www.cdc.gov/cholera/treatment/antibiotic-treatment.html (accessed on 15 October 2022).
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The Microbial Pan-Genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Hudzicki, J. Kirby-Bauer Disk Diffusion Susceptibility Test Protocol. Am. Soc. Microbiol. 2009, 15, 55–63. [Google Scholar]
- Prussing, C.; Snavely, E.A.; Singh, N.; Lapierre, P.; Lasek-Nesselquist, E.; Mitchell, K.; Haas, W.; Owsiak, R.; Nazarian, E.; Musser, K.A. Nanopore MinION Sequencing Reveals Possible Transfer of BlaKPC–2 Plasmid Across Bacterial Species in Two Healthcare Facilities. Front. Microbiol. 2020, 11, 2007. [Google Scholar] [CrossRef]
- Macqueen, D.J.; Eve, O.; Gundappa, M.K.; Daniels, R.R.; Gallagher, M.D.; Alexandersen, S.; Karlsen, M. Genomic Epidemiology of Salmonid Alphavirus in Norwegian Aquaculture Reveals Recent Subtype-2 Transmission Dynamics and Novel Subtype-3 Lineages. Viruses 2021, 13, 2549. [Google Scholar] [CrossRef]
- Cornet, L.; Baurain, D. Contamination Detection in Genomic Data: More Is Not Enough. Genome Biol. 2022, 23, 60. [Google Scholar] [CrossRef]
- Salvà-Serra, F.; Jaén-Luchoro, D.; Jakobsson, H.E.; Gonzales-Siles, L.; Karlsson, R.; Busquets, A.; Gomila, M.; Bennasar-Figueras, A.; Russell, J.E.; Fazal, M.A.; et al. Complete Genome Sequences of Streptococcus Pyogenes Type Strain Reveal 100%-Match between PacBio-Solo and Illumina-Oxford Nanopore Hybrid Assemblies. Sci. Rep. 2020, 10, 11656. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome Sequence-Based Species Delimitation with Confidence Intervals and Improved Distance Functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Joensen, K.G.; Zankari, E.; Ahrenfeldt, J.; Lukjancenko, O.; Kaas, R.S.; Roer, L.; Leekitcharoenphon, P.; Saputra, D.; Cosentino, S.; et al. Applied Genomics of Foodborne Pathogens. In Applied Genomics of Foodborne Pathogens; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the All-Bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- McDonald, N.D.; Regmi, A.; Morreale, D.P.; Borowski, J.D.; Boyd, E.F.; Fidelma Boyd, E. CRISPR-Cas Systems Are Present Predominantly on Mobile Genetic Elements in Vibrio Species. BMC Genom. 2019, 20, 105. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An Updated Database of Bacterial Integrative and Conjugative Elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The Reference Centre for Bacterial Insertion Sequences. Nucleic Acids Res. 2006, 34, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Galata, V.; Fehlmann, T.; Backes, C.; Keller, A. PLSDB: A Resource of Complete Bacterial Plasmids. Nucleic Acids Res. 2019, 47, D195–D202. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An Interactive Viewer for Bacterial Population Genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, I.; Chang, A.; Placzek, S.; Söhngen, C.; Rother, M.; Lang, M.; Munaretto, C.; Ulas, S.; Stelzer, M.; Grote, A.; et al. BRENDA in 2013: Integrated Reactions, Kinetic Data, Enzyme Function Data, Improved Disease Classification: New Options and Contents in BRENDA. Nucleic Acids Res. 2013, 41, D764–D772. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG Databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Ladrière, M. Current indications of azathioprine in nephrology. Nephrol. Ther. 2013, 9, 8–12. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; Waldor, M.K. Filamentous Phages Linked to Virulence of Vibrio Cholerae. Curr. Opin. Microbiol. 2003, 6, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Deb, S.; Arakawa, E.; Yamasaki, S.; Das, S.K. Pufferfish (Tetraodon Cutcutia) Sampled from a Freshwater River Serves as an Intermediate Reservoir of a Sucrose Nonfermenting Variant of Vibrio Cholerae PS-4. Microbiol. Spectr. 2022, 10, e0122121. [Google Scholar] [CrossRef] [PubMed]
- Weil, A.A.; Becker, R.L.; Harris, J.B. Vibrio Cholerae at the Intersection of Immunity and the Microbiome. MSphere 2019, 4, E00597-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, N.; Rivard, N.; Ceccarelli, D.; Colwell, R.R.; Burrus, V. IncA/C Conjugative Plasmids Mobilize a New Family of Multidrug Resistance Islands in Clinical Vibrio Cholerae Non-O1/Non-O139 Isolates from Haiti. mBio 2016, 7, e00509-16. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Verma, J.; Kumar, P.; Ghosh, A.; Ramamurthy, T. Antibiotic Resistance in Vibrio Cholerae: Understanding the Ecology of Resistance Genes and Mechanisms. Vaccine 2020, 38, A83–A92. [Google Scholar] [CrossRef]
- Marin, M.A.; Fonseca, E.L.; Andrade, B.N.; Cabral, A.C.; Vicente, A.C.P. Worldwide Occurrence of Integrative Conjugative Element Encoding Multidrug Resistance Determinants in Epidemic Vibrio Cholerae O1. PLoS ONE 2014, 9, e108728. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Lee, C.H.; Nair, G.B.; Kim, D.W. Whole-Genome Sequence Comparisons Reveal the Evolution of Vibrio Cholerae O1. Trends Microbiol. 2015, 23, 479–489. [Google Scholar] [CrossRef]
- Kim, E.J.; Lee, D.; Moon, S.H.; Lee, C.H.; Kim, D.W. CTX Prophages in Vibrio Cholerae O1 Strains. J. Microbiol. Biotechnol. 2014, 24, 725–731. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Price, L.B.; Schupp, J.M.; Gillece, J.D.; Kaas, R.S.; Engelthaler, D.M.; Bortolaia, V.; Pearson, T.; Waters, A.E.; Upadhyay, B.P.; et al. Population Genetics of Vibrio Cholerae from Nepal in 2010: Evidence on the Origin of the Haitian Outbreak. mBio 2011, 2, e00157-11. [Google Scholar] [CrossRef] [Green Version]
- Aruhomukama, D.; Sserwadda, I.; Mboowa, G. Whole-Genome Sequence Analysis of Vibrio Cholerae from Three Outbreaks in Uganda, 2014–2016. F1000Research 2019, 8, e0007330. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, V.M.; Gandhi, T.N.; Petty, L.A.; Patel, P.K.; Prescott, H.C.; Malani, A.N.; Ratz, D.; McLaughlin, E.; Chopra, V.; Flanders, S.A. Empiric Antibacterial Therapy and Community-Onset Bacterial Coinfection in Patients Hospitalized With Coronavirus Disease 2019 (COVID-19): A Multi-Hospital Cohort Study. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 72, e533–e541. [Google Scholar] [CrossRef] [PubMed]
- Marchaim, D.; Chopra, T.; Bhargava, A.; Bogan, C.; Dhar, S.; Hayakawa, K.; Pogue, J.M.; Bheemreddy, S.; Blunden, C.; Shango, M.; et al. Recent Exposure to Antimicrobials and Carbapenem-Resistant Enterobacteriaceae: The Role of Antimicrobial Stewardship. Infect. Control Hosp. Epidemiol. 2012, 33, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Ang, H.; Sun, X. Risk Factors for Multidrug-resistant Gram-negative Bacteria Infection in Intensive Care Units: A Meta-analysis. Int. J. Nurs. Pract. 2018, 24, e12644. [Google Scholar] [CrossRef] [PubMed]
- Chedid, M.; Waked, R.; Haddad, E.; Chetata, N.; Saliba, G.; Choucair, J. Antibiotics in Treatment of COVID-19 Complications: A Review of Frequency, Indications, and Efficacy. J. Infect. Public Health 2021, 14, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Borgeaud, S.; Metzger, L.C.; Scrignari, T.; Blokesch, M. The Type VI Secretion System of Vibrio Cholerae Fosters Horizontal Gene Transfer. Science 2015, 347, 63–67. [Google Scholar] [CrossRef]
- Antonova, E.S.; Hammer, B.K. Quorum-Sensing Autoinducer Molecules Produced by Members of a Multispecies Biofilm Promote Horizontal Gene Transfer to Vibrio Cholerae. FEMS Microbiol. Lett. 2011, 322, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Mohanraj, R.S.; Mandal, J. Azithromycin Can Induce SOS Response and Horizontal Gene Transfer of SXT Element in Vibrio Cholerae. Mol. Biol. Rep. 2022, 49, 4737–4748. [Google Scholar] [CrossRef]
- Verma, J.; Bag, S.; Saha, B.; Kumar, P.; Ghosh, T.S.; Dayal, M.; Senapati, T.; Mehra, S.; Dey, P.; Desigamani, A.; et al. Genomic Plasticity Associated with Antimicrobial Resistance in Vibrio Cholerae. Proc. Natl. Acad. Sci. USA 2019, 116, 6226–6231. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, C.; Grohmann, E. Horizontal Gene Transfer of Antibiotic Resistance Genes in Biofilms. Antibiotics 2023, 12, 328. [Google Scholar] [CrossRef]
- Igbinosa, E.O.; Obi, L.C.; Tom, M.; Okoh, A.I. Detection of Potential Risk of Wastewater Effluents for Transmission of Antibiotic Resistance from Vibrio Species as a Reservoir in a Peri-Urban Community in South Africa. Int. J. Environ. Health Res. 2011, 21, 402–414. [Google Scholar] [CrossRef]
- Maharajan, A.D.; Hjerde, E.; Hansen, H.; Willassen, N.P. Quorum Sensing Controls the CRISPR and Type VI Secretion Systems in Aliivibrio Wodanis 06/09/139. Front. Vet. Sci. 2022, 9, 799414. [Google Scholar] [CrossRef]
- Maruyama, A.; Kumagai, Y.; Morikawa, K.; Taguchi, K.; Hayashi, H.; Ohta, T. Oxidative-Stress-Inducible QorA Encodes an NADPH-Dependent Quinone Oxidoreductase Catalysing a One-Electron Reduction in Staphylococcus Aureus. Microbiology 2003, 149, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Reytor, D.; Pavón, A.; Lopez-Joven, C.; Ramírez-Araya, S.; Peña-Varas, C.; Plaza, N.; Alegría-Arcos, M.; Corsini, G.; Jaña, V.; Pavez, L.; et al. Analysis of the Zonula Occludens Toxin Found in the Genome of the Chilean Non-Toxigenic Vibrio Parahaemolyticus Strain PMC53.7. Front. Cell. Infect. Microbiol. 2020, 10, 482. [Google Scholar] [CrossRef]
- DeAngelis, C.M.; Nag, D.; Withey, J.H.; Matson, J.S. Characterization of the Vibrio Cholerae Phage Shock Protein Response. J. Bacteriol. 2019, 201, e00761-18. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.P.; Guay, G.G. Gene Amplification Contributes to Sulfonamide Resistance in Escherichia Coli. Antimicrob. Agents Chemother. 1989, 33, 2042–2048. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.E.; Smith, M.; Wilkinson, M.C.; Peek, K. Identification and Characterization of a Chitinase Antigen from Pseudomonas Aeruginosa Strain 385. Appl. Environ. Microbiol. 2001, 67, 4001–4008. [Google Scholar] [CrossRef] [Green Version]
- De, R. Mobile Genetic Elements of Vibrio Cholerae and the Evolution of Its Antimicrobial Resistance. Front. Trop. Dis. 2021, 2, 691604. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, H.J.; Lee, S.J. Complete Genome Sequence of Escherichia Coli Strain BL21. Genome Announc. 2015, 3, e00134-15. [Google Scholar] [CrossRef] [Green Version]
- Colavecchio, A.; D’Souza, Y.; Tompkins, E.; Jeukens, J.; Freschi, L.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Boyle, B.; Bekal, S.; Tamber, S.; et al. Prophage Integrase Typing Is a Useful Indicator of Genomic Diversity in Salmonella Enterica. Front. Microbiol. 2017, 8, 1283. [Google Scholar] [CrossRef] [Green Version]
- De Jonge, N.; Hohlweg, W.; Garcia-Pino, A.; Respondek, M.; Buts, L.; Haesaerts, S.; Lah, J.; Zangger, K.; Loris, R. Structural and Thermodynamic Characterization of Vibrio Fischeri CcdB. J. Biol. Chem. 2010, 285, 5606–5613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehours, P.; Freydière, A.-M.; Richer, O.; Burucoa, C.; Boisset, S.; Lanotte, P.; Prère, M.F.; Ferroni, A.; Lafuente, C.; Vandenesch, F.; et al. The RtxA Toxin Gene of Kingella Kingae: A Pertinent Target for Molecular Diagnosis of Osteoarticular Infections. J. Clin. Microbiol. 2011, 49, 1245–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, B.D.; Patel, D.; Seo, S.M.; Kaatz, G.W. Mutagenesis and Modeling to Predict Structural and Functional Characteristics of the Staphylococcus Aureus MepA Multidrug Efflux Pump. J. Bacteriol. 2013, 195, 523–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, B.P.; Davis, B.M.; Jie, Y.; Waldor, M.K. Characterization of a HigBA Toxin-Antitoxin Locus in Vibrio Cholerae. J. Bacteriol. 2007, 189, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Ohki, R.; Murata, M. Bmr3, a Third Multidrug Transporter Gene of Bacillus Subtilis. J. Bacteriol. 1997, 179, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Crosby, J.A.; Kachlany, S.C. TdeA, a TolC-like Protein Required for Toxin and Drug Export in Aggregatibacter (Actinobacillus) Actinomycetemcomitans. Gene 2007, 388, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Mudrak, B.; Tamayo, R. The Vibrio Cholerae Pst2 Phosphate Transport System Is Upregulated in Biofilms and Contributes to Biofilm-Induced Hyperinfectivity. Infect. Immun. 2012, 80, 1794–1802. [Google Scholar] [CrossRef] [Green Version]
- Foote, S.J.; Bossé, J.T.; Bouevitch, A.B.; Langford, P.R.; Young, N.M.; Nash, J.H.E. The Complete Genome Sequence of Actinobacillus Pleuropneumoniae L20 (Serotype 5b). J. Bacteriol. 2008, 190, 1495–1496. [Google Scholar] [CrossRef] [Green Version]
- Amitai, S.; Yassin, Y.; Engelberg-Kulka, H. MazF-Mediated Cell Death in Escherichia Coli: A Point of No Return. J. Bacteriol. 2004, 186, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Moch, C.; Fromant, M.; Blanquet, S.; Plateau, P. DNA Binding Specificities of Escherichia Coli Cas1-Cas2 Integrase Drive Its Recruitment at the CRISPR Locus. Nucleic Acids Res. 2017, 45, 2714–2723. [Google Scholar] [CrossRef] [Green Version]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia Coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-Y.; Ma, L.-Y.; Yu, L.; Lu, X.; Liang, W.-L.; Kan, B.; Su, J.-R. Quinolone Resistance Genes and Their Contribution to Resistance in Vibrio Cholerae Serogroup O139. Antibiotics 2023, 12, 416. [Google Scholar] [CrossRef]
- Nohejl, T.; Valcek, A.; Papousek, I.; Palkovicova, J.; Wailan, A.M.; Pratova, H.; Minoia, M.; Dolejska, M. Genomic Analysis of Qnr-Harbouring IncX Plasmids and Their Transferability within Different Hosts under Induced Stress. BMC Microbiol. 2022, 22, 136. [Google Scholar] [CrossRef]
- Morita, D.; Takahashi, E.; Morita, M.; Ohnishi, M.; Mizuno, T.; Miyoshi, S.; Dutta, D.; Ramamurthy, T.; Chowdhury, G.; Mukhopadhyay, A.K.; et al. Genomic Characterization of Antibiotic Resistance-Encoding Genes in Clinical Isolates of Vibrio Cholerae Non-O1/Non-O139 Strains from Kolkata, India: Generation of Novel Types of Genomic Islands Containing Plural Antibiotic Resistance Genes. Microbiol. Immunol. 2020, 64, 435–444. [Google Scholar] [CrossRef]
- Mogasale, V.; Levin, A.; Maskery, B.; DeRoeck, D.; Kim, Y.E.; Clemens, J.; Lopez, A.L.; Burgess, C.; Wierzba, T. Oral Cholera Vaccines to Control Endemic Disease: An Economic and Epidemiological Modelling Analysis. Lancet 2013, 382, 6. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
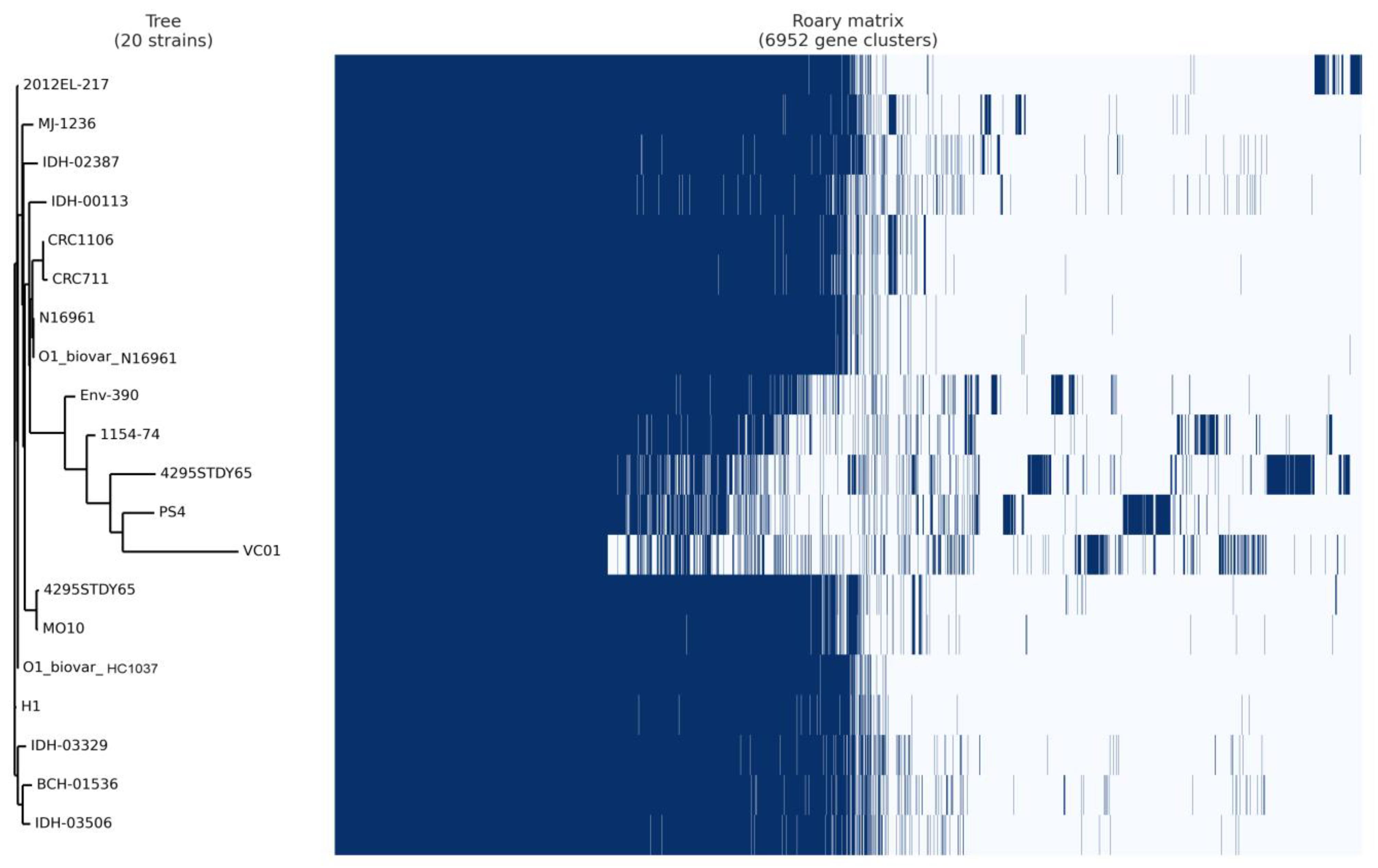{kind=link}
| Locus | Identity | Coverage | Alignment Length | Allele Length | Gaps | Organism |
|---|---|---|---|---|---|---|
| adk | 100 | 100 | 416 | 416 | 0 | V. cholerae |
| gyrB | 99.768 | 99.768 | 431 | 431 | 1 | |
| mdh | 99.0499 | 99.0499 | 421 | 421 | 4 | |
| metE | 100 | 100 | 591 | 591 | 0 | |
| pntA | 100 | 100 | 431 | 431 | 0 | |
| purM | 99.5798 | 99.5798 | 476 | 476 | 2 | |
| pyrC | 99.5546 | 100 | 449 | 449 | 0 |
| No | Protein Feature | Count |
|---|---|---|
| 1 | Hypothetical proteins | 812 |
| 2 | Proteins with functional assignments | 4870 |
| 3 | Proteins with EC number assignments | 1614 |
| 4 | Proteins with GO assignments | 1324 |
| 5 | Proteins with Pathway assignments | 1136 |
| 6 | Proteins with PATRIC genus-specific family (PLfam) assignments | 5486 |
| 7 | Proteins with PATRIC cross-genus family (PGfam) assignments | 5579 |
| Resistance Mechanism | Gene | AMR Gene Family | Drug Class | Identity (%) | Coverage (%) |
|---|---|---|---|---|---|
| Antibiotic efflux | CRP | resistance-nodulation-cell division (RND) antibiotic efflux pump | macrolide antibiotic, fluoroquinolone antibiotic, penam | 95.24 | 100 |
| Antibiotic inactivation | APH (3″)-Ib | APH (3″) | aminoglycoside | 98.81 | 94.38 |
| catB9 | chloramphenicol acetyltransferase (CAT) | phenicol antibiotic | 99.52 | 99.52 | |
| Vibrio cholerae varG | Subclass B1 Vibrio cholerae varG beta-lactamase | carbapenem | 99.7 | 84.87 | |
| Antibiotic target alteration | E. coli EF-Tu | elfamycin resistant EF-Tu | elfamycin antibiotic | 87.28 | 96.33 |
| dfrA1 | trimethoprim-resistant dihydrofolate reductase dfr | diaminopyrimidine | 99.36 | 100 | |
| Antibiotics resistance | SUL2, DFRA1 | dihydropteroate synthase (DHPS) | Sulfonamide and trimethoprim-resistant dihydropteroate synthase Sul2 | 99.14 | 98.9 |
| Efflux Regulator | EMRD | Drug and biocide resistance | Multidrug efflux | 100 | 100 |
| VCEA | Drug and biocide resistance | Multidrug efflux | 98.24 | 99.75 | |
| VCEB | Drug and biocide resistance | Multidrug efflux | 98.31 | 99.93 | |
| VCER | Drug and biocide resistance | Multidrug efflux | 99.83 | 99.83 | |
| VCMA | Drug and biocide resistance | Multidrug efflux | 99.71 | 99.86 |
| Region | Length | Completeness | Score | Total Proteins | Region Position | Most Common Phage | GC (%) |
|---|---|---|---|---|---|---|---|
| 1 | 10.7 Kb | incomplete | 60 | 14 | 315,989–326,748 | PHAGE_Escher_500465_1_ NC_049342(7) | 47.13 |
| 2 | 12.1 Kb | intact | 150 | 30 | 966,779–978,912 | PHAGE_Vibrio_CTX_ NC_015209(13) | 45.67 |
| 3 | 12.7 Kb | incomplete | 50 | 26 | 1,339,522–1,352,283 | PHAGE_Pseudo_JBD18_ NC_027986(3) | 42.96 |
| 4 | 20.6 Kb | incomplete | 60 | 13 | 3,749,246–3,769,881 | PHAGE_Escher_RCS47_ NC_042128(2) | 45.76 |
| No | Cluster | Gene | Annotation | Function | Organism | Reference |
|---|---|---|---|---|---|---|
| 1 | casC | casC | CRISPR system Cascade subunit CasC | CRISPR | Vibrio spp. | [29] |
| 2 | csy3 | csy3 | CRISPR-associated protein Csy3 | CRISPR | Aliivibrio wodanis | [64] |
| 3 | group_1026 | qorA | Quinone oxidoreductase 1 | Resistance | Staphylococcus aureus | [65] |
| 4 | group_1096 | zot | Zona occludens toxin | Toxin | Vibrio parahaemolyticus | [66] |
| 5 | group_1408 | pspA | Phage shock protein A | Phage | Vibrio cholerae | [67] |
| 6 | group_1432 | bcr | Bicyclomycin resistance protein | Resistance | Escherichia coli K12 | [68] |
| 7 | group_146 | endo I | Chitodextrinase | Resistance | Pseudoalteromonas sp. P1-9 | [69] |
| 8 | group_1966 | tcpE | Toxin coregulated pilus biosynthesis protein E | Toxin | Vibrio cholerae | [70] |
| 9 | group_2021 | group_2021 | IS3 family transposase ISVpa4 | Resistance | Escherichia coli | [71] |
| 10 | group_307 | intA | Prophage integrase IntA | Phage | Salmonella enterica | [72] |
| 11 | group_3154 | ccdB | Toxin CcdB | Toxin | Vibrio fischeri | [73] |
| 12 | group_33 | rtxA | Multifunctional-autoprocessing repeats-in-toxin | Toxin | Kingella kingae | [74] |
| 13 | group_373 | mepA | Multidrug export protein MepA | Resistance | Staphylococcus aureus | [75] |
| 14 | group_3960 | higB-1 | Toxin HigB-1 | Toxin | Vibrio cholerae | [76] |
| 15 | group_3962 | higA-1 | Antitoxin HigA-1 | Toxin | [76] | |
| 16 | group_442 | bmr3 | Multidrug resistance protein 3 | Resistance | Bacillus subtilis | [77] |
| 17 | group_461 | ltxB | Leukotoxin export ATP-binding protein LtxB | Toxin | Aggregatibacter actinomycetemcomitans | [78] |
| 18 | group_655 | fruB | Multiphosphoryl transfer protein | Resistance | Vibrio cholerae Pst2 | [79] |
| 19 | group_76 | apxIB | Toxin RTX-I translocation ATP-binding protein | Toxin | Actinobacillus pleuropneumoniae | [80] |
| 20 | mazF | mazF | Endoribonuclease toxin MazF | Toxin | Escherichia coli | [81] |
| 21 | ygbF | ygbF | CRISPR-associated endoribonuclease Cas2 | CRISPR | Escherichia coli | [82] |
| 22 | ygbT | ygbT | CRISPR-associated endonuclease Cas1 | CRISPR | Escherichia coli K12 | [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mevada, V.; Patel, R.; Dudhagara, P.; Chaudhari, R.; Vohra, M.; Khan, V.; J. H. Shyu, D.; Chen, Y.-Y.; Zala, D. Whole Genome Sequencing and Pan-Genomic Analysis of Multidrug-Resistant Vibrio cholerae VC01 Isolated from a Clinical Sample. Microorganisms 2023, 11, 2030. https://doi.org/10.3390/microorganisms11082030
Mevada V, Patel R, Dudhagara P, Chaudhari R, Vohra M, Khan V, J. H. Shyu D, Chen Y-Y, Zala D. Whole Genome Sequencing and Pan-Genomic Analysis of Multidrug-Resistant Vibrio cholerae VC01 Isolated from a Clinical Sample. Microorganisms. 2023; 11(8):2030. https://doi.org/10.3390/microorganisms11082030
Chicago/Turabian StyleMevada, Vishal, Rajesh Patel, Pravin Dudhagara, Rajesh Chaudhari, Mustafa Vohra, Vikram Khan, Douglas J. H. Shyu, Yih-Yuan Chen, and Dolatsinh Zala. 2023. "Whole Genome Sequencing and Pan-Genomic Analysis of Multidrug-Resistant Vibrio cholerae VC01 Isolated from a Clinical Sample" Microorganisms 11, no. 8: 2030. https://doi.org/10.3390/microorganisms11082030
APA StyleMevada, V., Patel, R., Dudhagara, P., Chaudhari, R., Vohra, M., Khan, V., J. H. Shyu, D., Chen, Y. -Y., & Zala, D. (2023). Whole Genome Sequencing and Pan-Genomic Analysis of Multidrug-Resistant Vibrio cholerae VC01 Isolated from a Clinical Sample. Microorganisms, 11(8), 2030. https://doi.org/10.3390/microorganisms11082030