Metagenomic Insights and Genomic Analysis of Phosphogypsum and Its Associated Plant Endophytic Microbiomes Reveals Valuable Actors for Waste Bioremediation


 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Phosphogypsum, and Effluents Collection
2.2. SF, SM, MN and AI Plant Materials Collection
2.3. DNA Extraction from Phosphogypsum and Plant Materials and Metagenomic Analysis
2.4. Isolation of Cultivable Bacterial Microbiome of Phosphogypsum and AI, SF, SM and MN Plants
2.5. Measurement of PGP Activities of Plant Endophytic Communities
2.6. Screening of Phosphogypsum and Plant Endophytic Bacterial Microbiomes for Antibiotic, Metal and Salinity (NaCl) Resistance, Pesticides and Effluents Degradation
2.7. Bacterial Genome Sequencing, Assembly and Annotation
2.8. Selection and Phylogenomic Analysis of Phosphogypsum Bacterial Isolates
2.9. Homology-based Mining of Genes Contributing to Detoxification of Organic Pollutants, Competition, Fitness and Comparative Genomics Analysis of BA Isolates
2.10. Secondary Metabolite Clusters Identification Using antiSMASH, NapDos, NP.Search, and Bagel3
2.11. Statistical Analysis
3. Results
3.1. Physicochemical Characteristics of Phosphogypsum
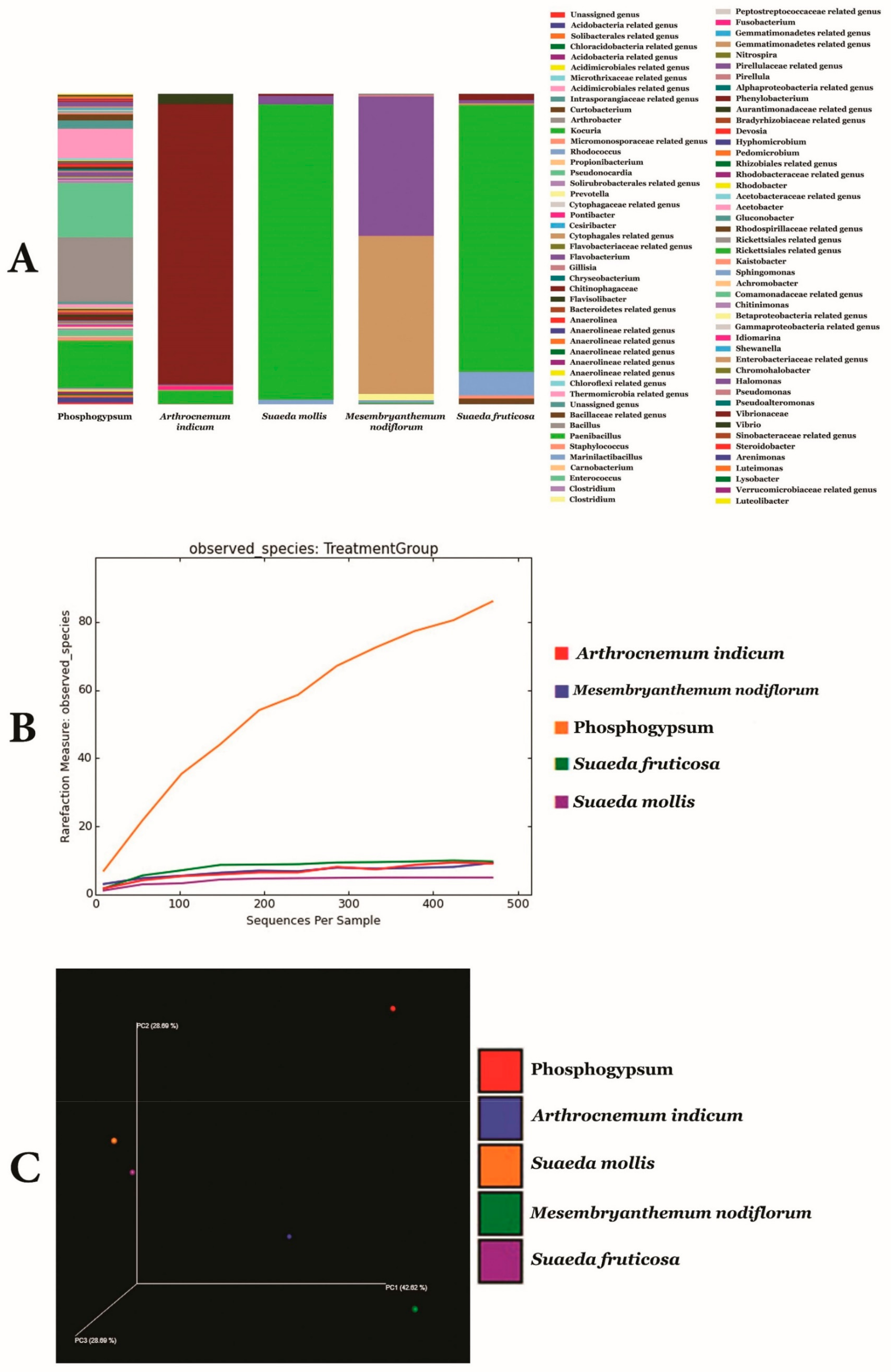
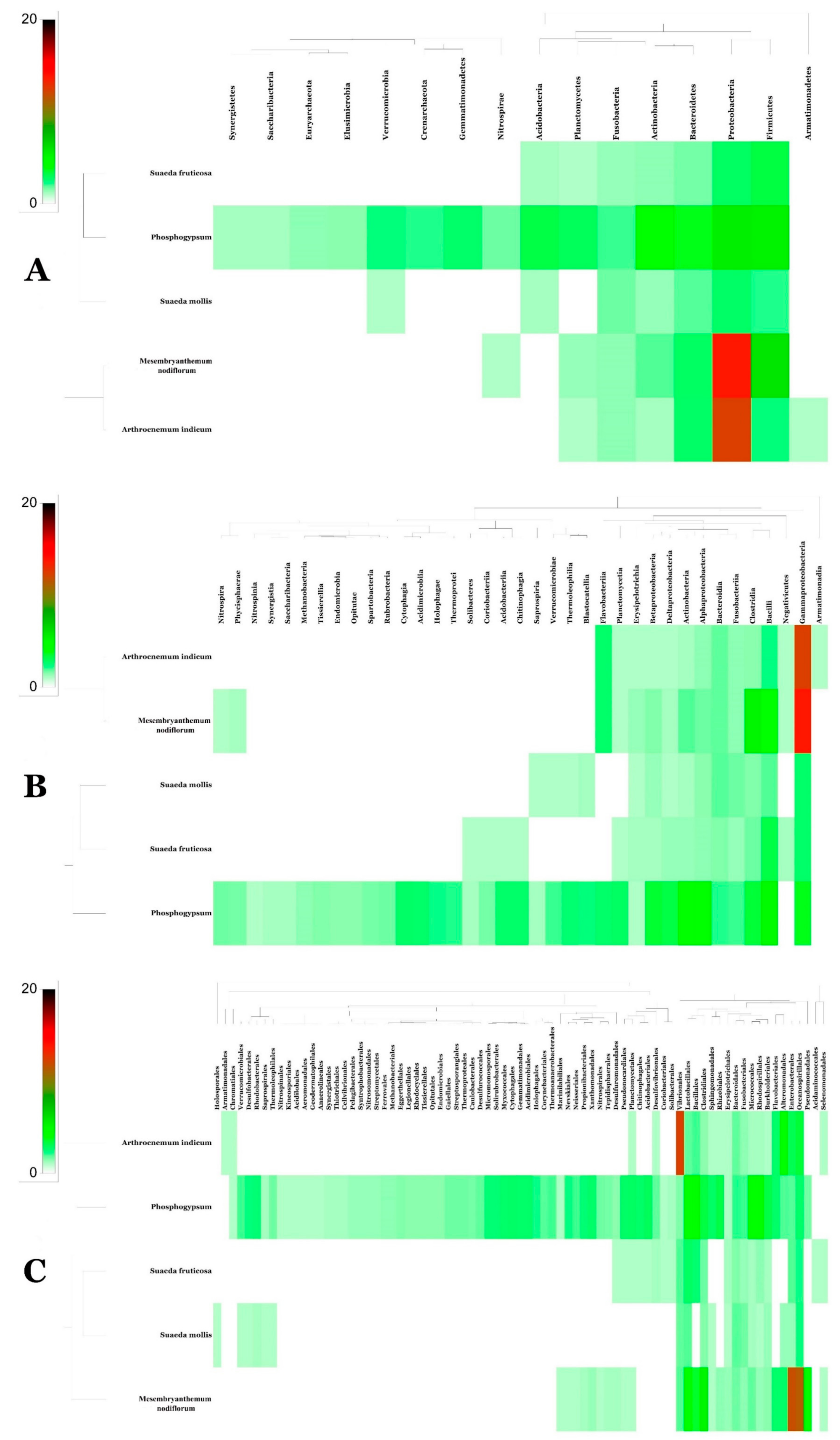
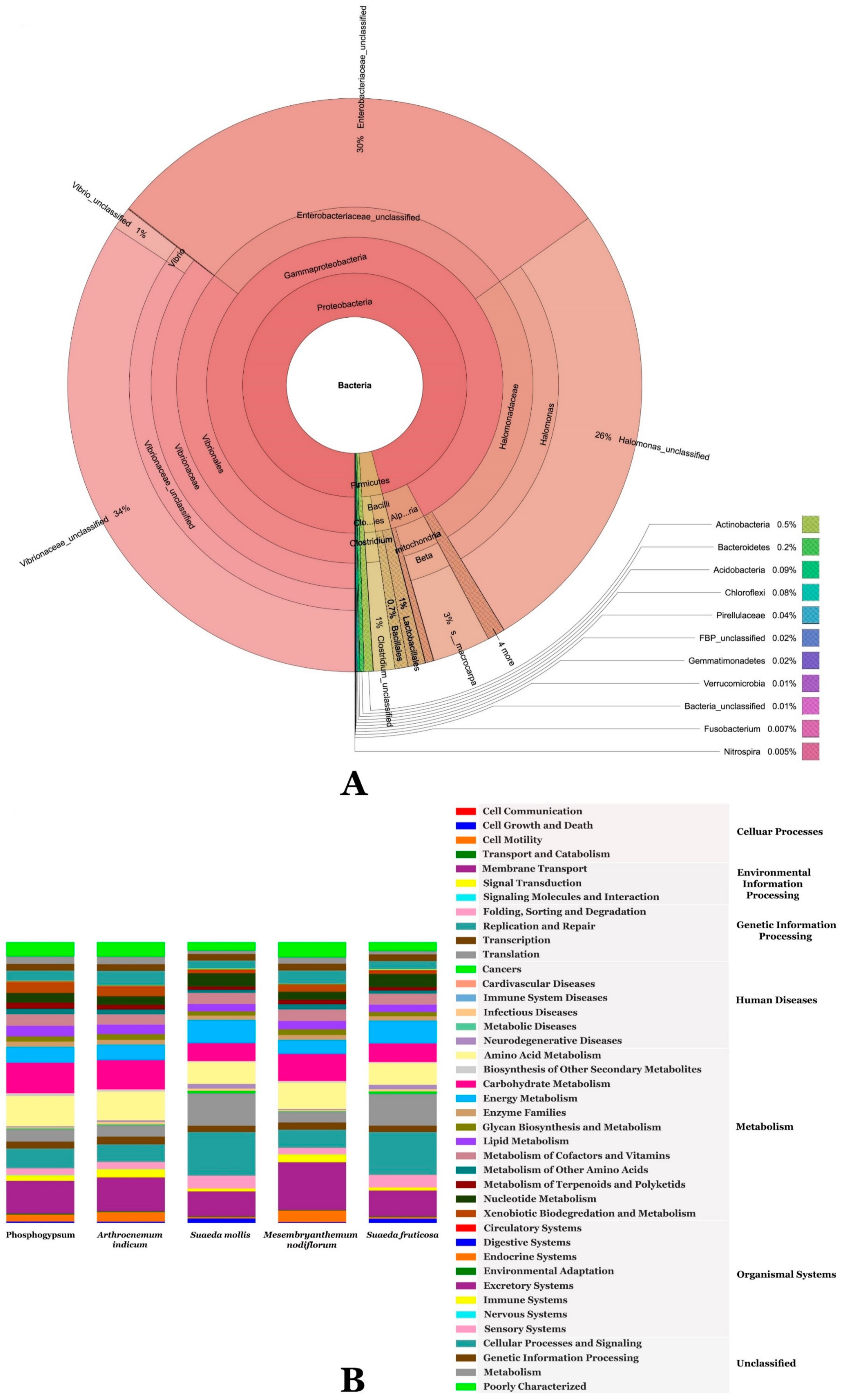
3.2. Metagenomic Analysis of Phosphogypsum and Plant Endophytic Microbiomes
3.3. Metagenomic Functional Content of Phosphogypsum and Plant Endophytic Microbiomes
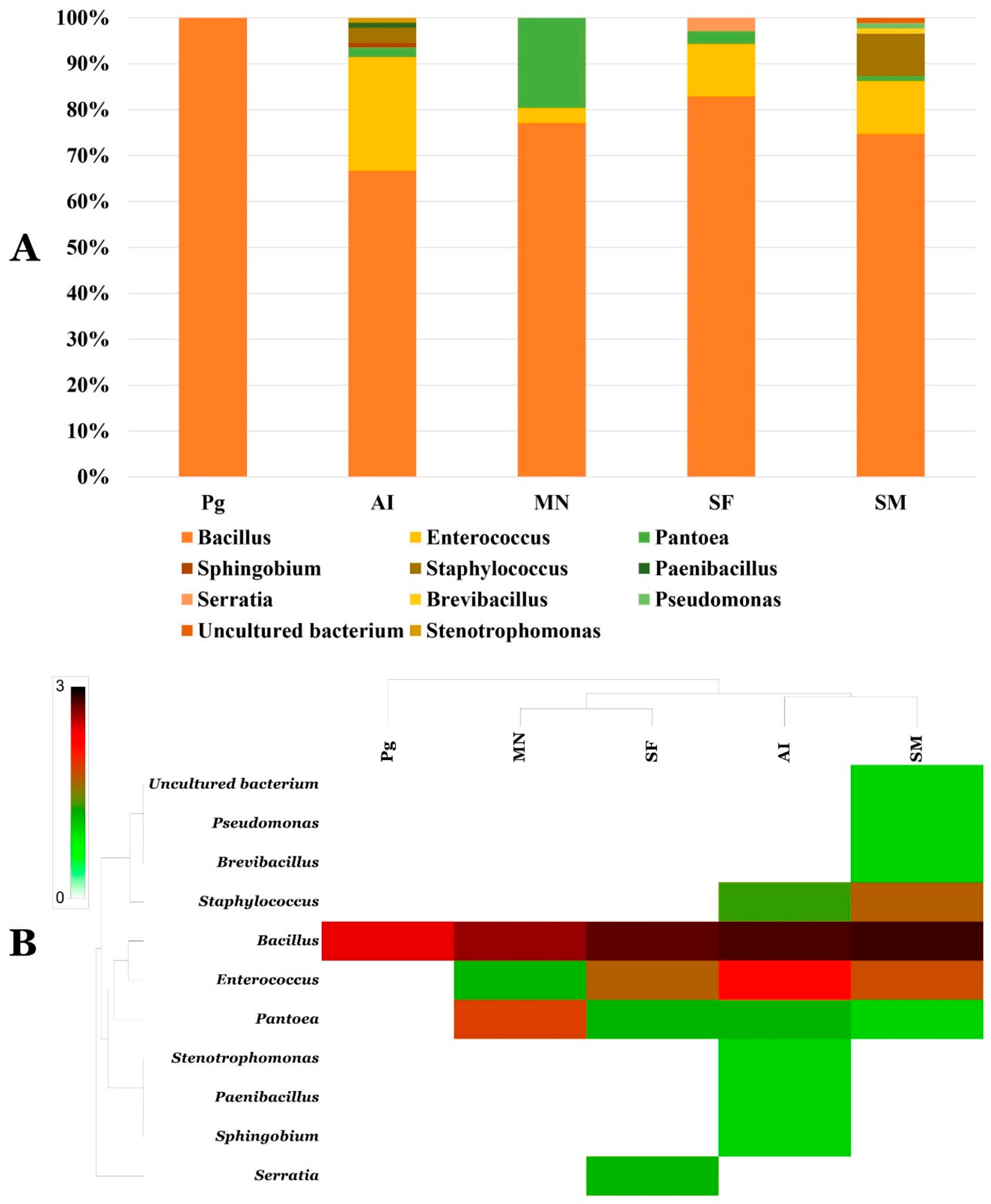
3.4. Phylogenetic Analysis of Cultivable Microbiomes of Phosphogypsum and Plant Endophytes
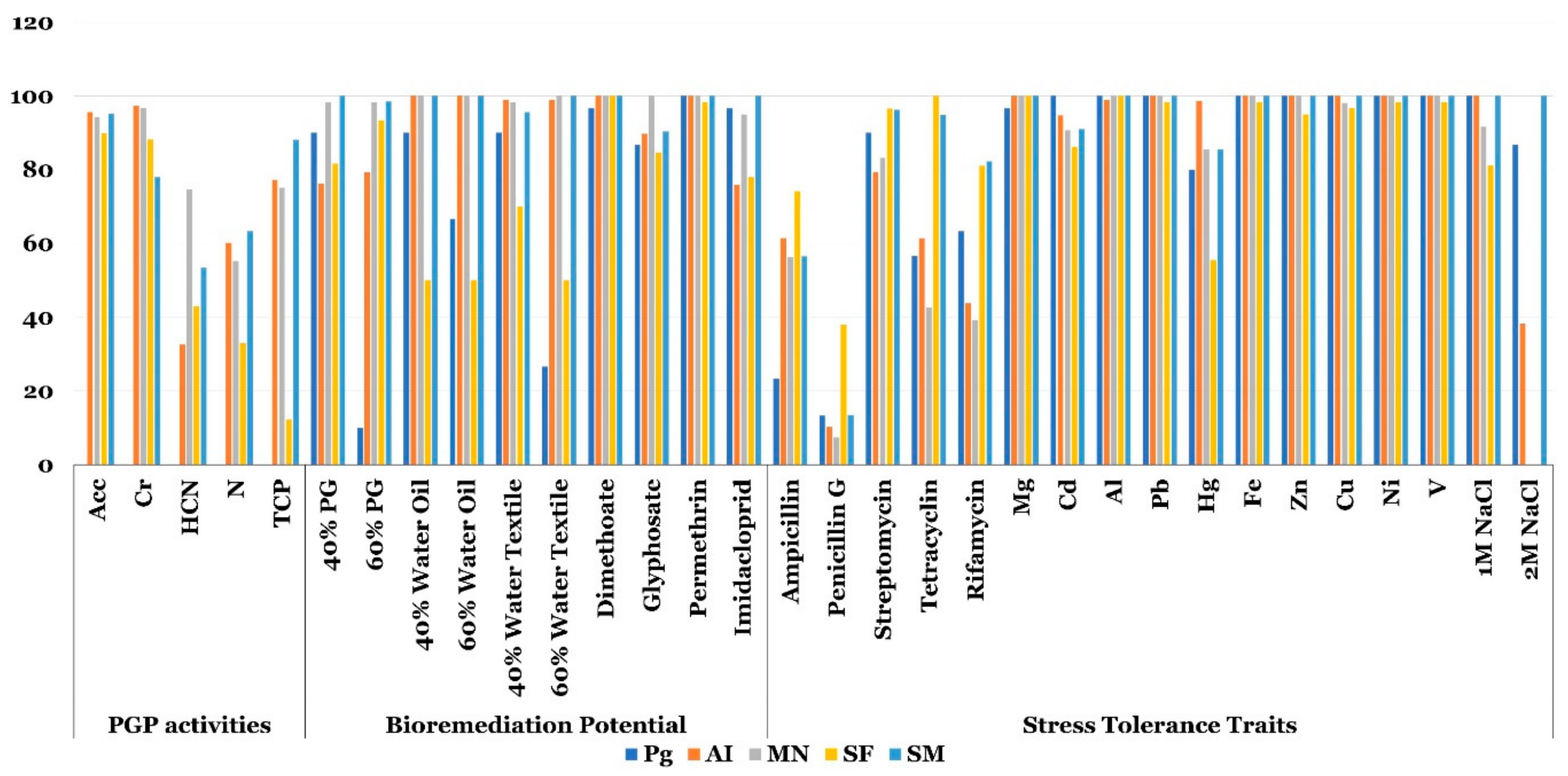
3.5. Characterization of Phosphogypsum Bacterial and Plant Endophytic Microbiomes for PGP Activities, Bioremediation Potential and Stress Tolerance
3.6. Identity and Phylogenomic Positions of Bacterial Phosphogypsum Isolates PG 1, PG 9, PG 17, PG 18 and PG 26
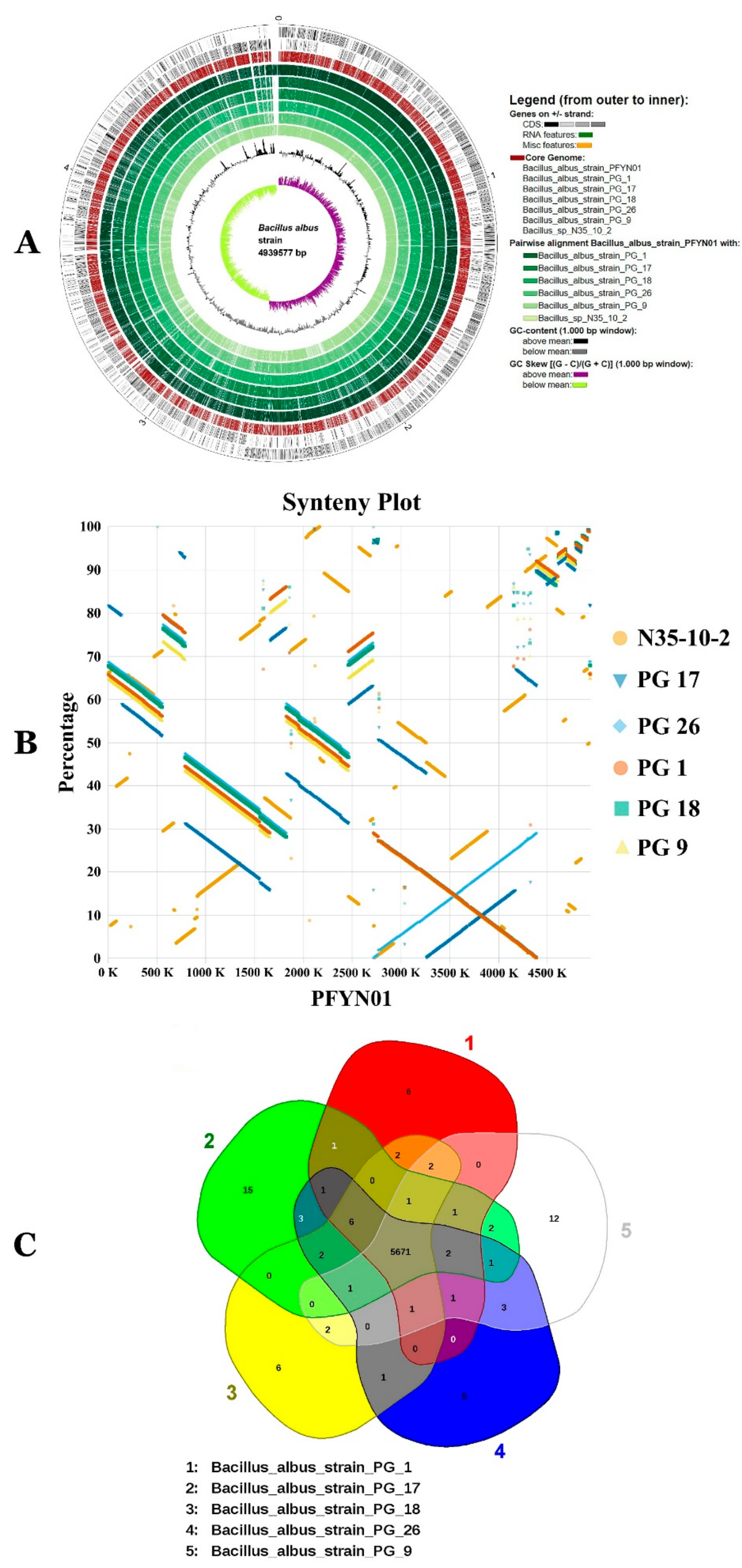
3.7. Characterization of the Core and Pan Genome of BA
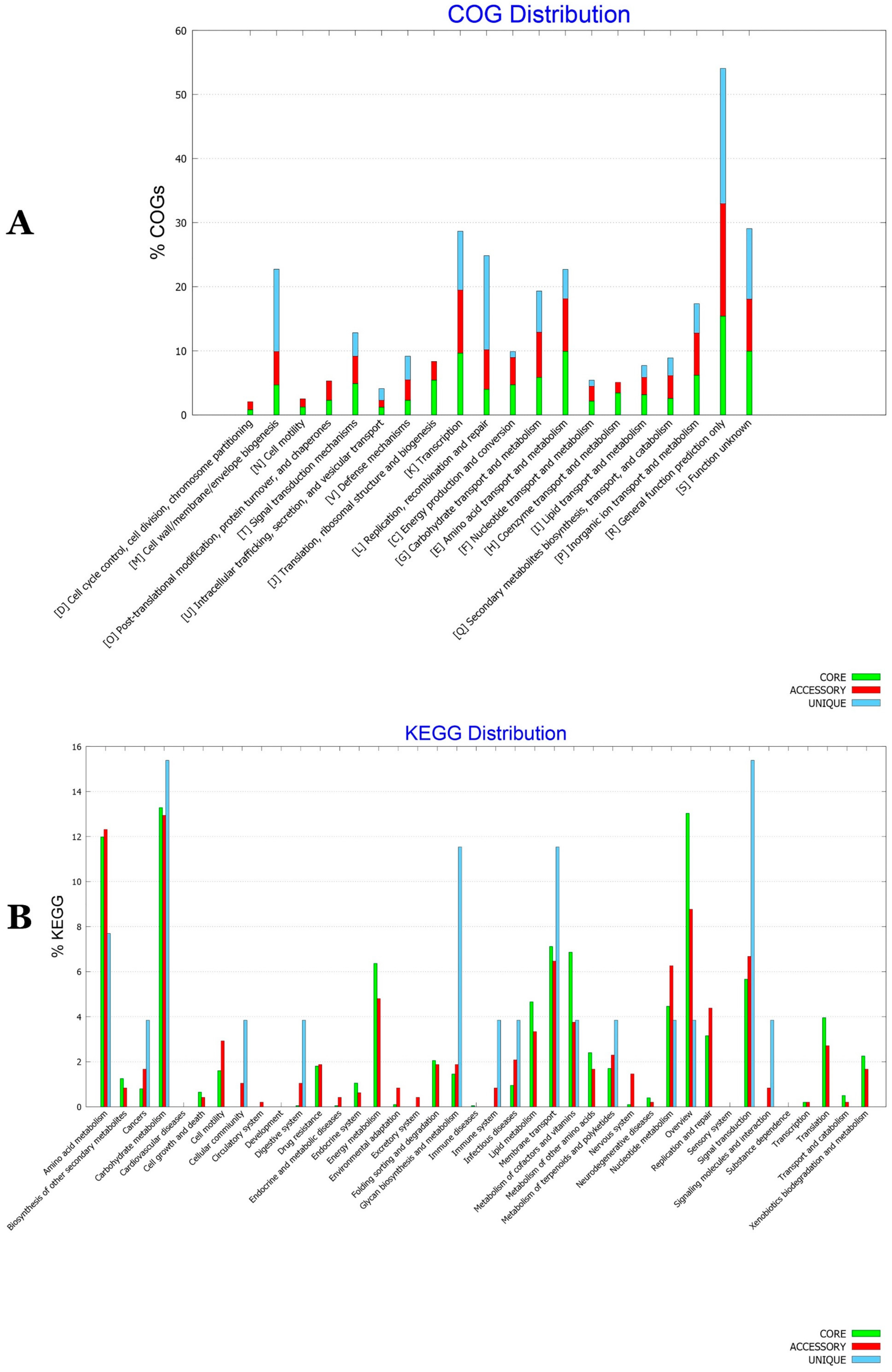
3.8. Functional Characterization of the BA Core, Accessory and Unique Genomes
3.9. SM Biosynthesis Abilities of the BA Pan, Core and Accessory Genomes
3.10. Predicted Natural Products Richness and Location within BA Genomes
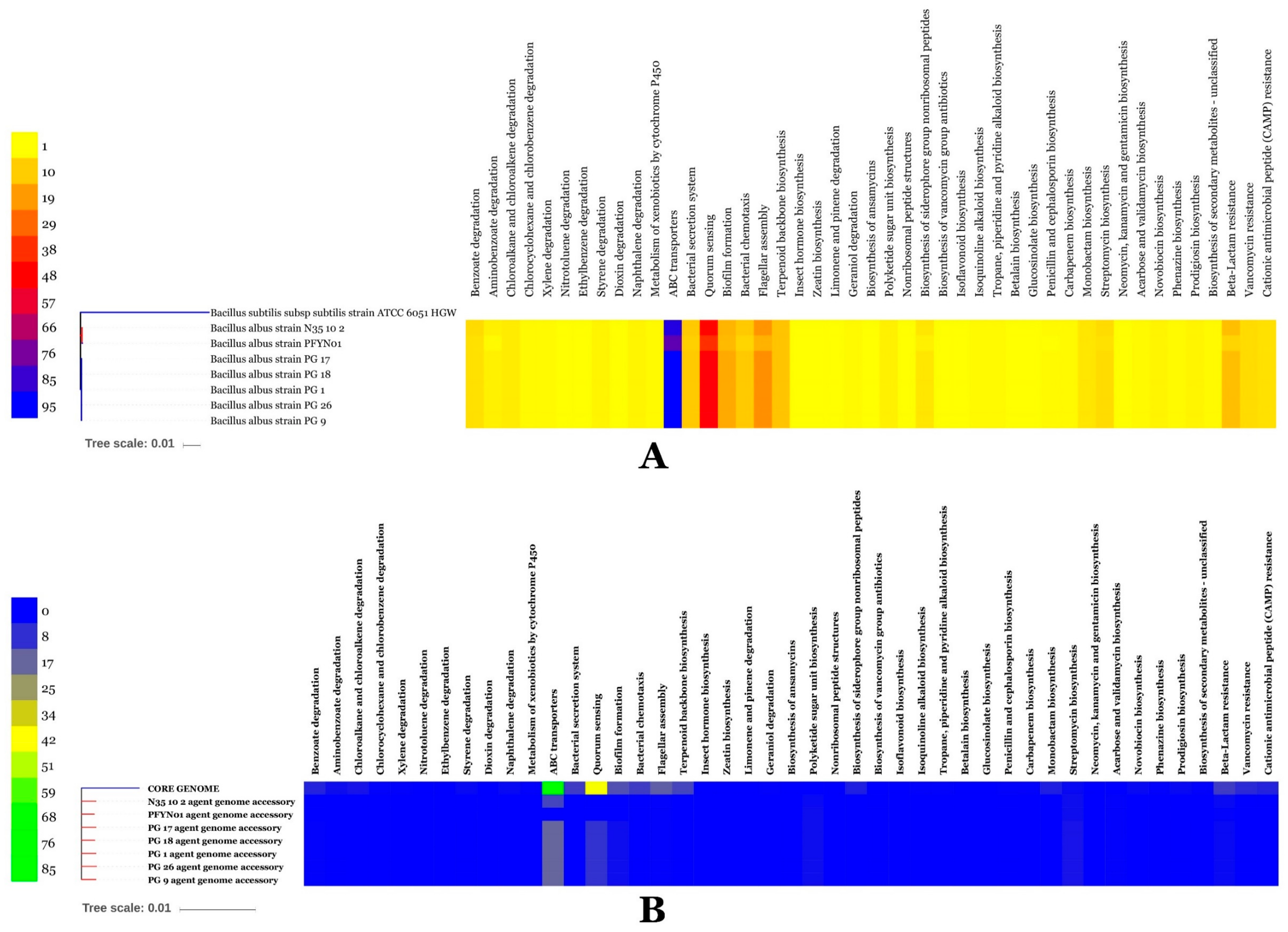
3.11. BA Genome Mining of Functional Genes for Detoxification of Organic Pollutants, Fitness and Competition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pérez-Moreno, S.M.; Gázquez, M.J.; Pérez-López, R.; Vioque, I.; Bolívar, J.P. Assessment of natural radionuclides mobility in a phosphogypsum disposal area. Chemosphere 2018, 211, 775–783. [Google Scholar] [CrossRef] [PubMed]
- El Kateb, A.; Stalder, C.; Rüggeberg, A.; Neururer, C.; Spangenberg, J.E.; Spezzaferri, S. Impact of industrial phosphate waste discharge on the marine environment in the Gulf of Gabes (Tunisia). PLoS ONE 2018, 13, e0197731. [Google Scholar] [CrossRef] [PubMed]
- Elloumi, N.; Zouari, M.; Chaari, L.; Abdallah, F.B.; Woodward, S.; Kallel, M. Effect of phosphogypsum on growth, physiology, and the antioxidative defense system in sunflower seedlings. Environ. Sci. Pollut. Res. 2015, 22, 14829–14840. [Google Scholar] [CrossRef] [PubMed]
- Hentati, O.; Abrantes, N.; Caetano, A.L.; Bouguerra, S.; Gonçalves, F.; Römbke, J.; Pereira, R. Phosphogypsum as a soil fertilizer: Ecotoxicity of amended soil and elutriates to bacteria, invertebrates, algae and plants. J. Hazard. Mater. 2015, 294, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Smaoui-Jardak, M.; Kriaa, W.; Maalej, M.; Zouari, M.; Kamoun, L.; Trabelsi, W.; Ben Abdallah, F.; Elloumi, N. Effect of the phosphogypsum amendment of saline and agricultural soils on growth, productivity and antioxidant enzyme activities of tomato (Solanum lycopersicum L.). Ecotoxicology 2017, 8, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Mishra, C.S.K.; Guru, B.C.; Samal, S. Histological anomalies and alterations in enzyme activities of the earthworm Glyphidrillus tuberosus exposed to high concentrations of phosphogypsum. Environ. Monit. Assess. 2018, 190, 529. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, H.; Karray, F.; Bartoli, M.; Ben Hnia, W.; Mhiri, N.; Fardeau, M.L.; Bennour, F.; Kamoun, L.; Alazard, D.; Sayadi, S. Desulfobulbus aggregans sp. nov., a novel sulfate reducing bacterium isolated from marine sediment from the gulf of Gabes. Curr. Microbiol. 2017, 74, 449–454. [Google Scholar] [CrossRef]
- Zouch, H.; Karray, F.; Armougom, F.; Chifflet, S.; Hirschler-Réa, A.; Kharrat, H.; Kamoun, L.; Ben Hania, W.; Ollivier, B.; Sayadi, S.; et al. Microbial diversity in sulfate-reducing marine sediment enrichment cultures associated with anaerobic biotransformation of coastal stockpiled Phosphogypsum (Sfax, Tunisia). Front. Microbiol. 2017, 8, 1583. [Google Scholar] [CrossRef]
- Alenezi, F.N.; Rekik, I.; Belka, M.; Ibrahim, A.F.; Luptakova, L.; Jaspars, M.; Woodward, S.; Belbahri, L. Strain-level diversity of secondary metabolism in the biocontrol species Aneurinibacillus migulanus. Microbiol. Res. 2016, 182, 116–124. [Google Scholar] [CrossRef]
- Alenezi, F.N.; Rekik, I.; Chenari Bouket, A.; Luptakova, L.; Weitz, H.J.; Rateb, M.E.; Jaspars, M.; Woodward, S.; Belbahri, L. Increased biological activity of Aneurinibacillus migulanus strains correlates with the production of new gramicidin secondary metabolites. Front. Microbiol 2017, 8, 517. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Khare, E.; Mishra, J.; Arora, N.K. Multifaceted interactions between endophytes and plant: Developments and prospects. Front. Microbiol. 2018, 15, 2732. [Google Scholar] [CrossRef] [PubMed]
- Mefteh, F.B.; Daoud, A.; Chenari Bouket, A.; Alenezi, F.N.; Luptakova, L.; Rateb, M.E.; Kadri, A.; Gharsallah, N.; Belbahri, L. Fungal root microbiome from healthy and brittle leaf diseased date palm trees (Phoenix dactylifera L.) reveals a hidden untapped arsenal of antibacterial and broad spectrum antifungal secondary metabolites. Front. Microbiol. 2017, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Young, E.; Carey, M.; Andrew, A.; Meharg, A.A.; Meharg, C. Microbiome and ecotypic adaption of Holcus lanatus (L.) to extremes of its soil pH range, investigated through transcriptome sequencing. Microbiome 2018, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Slama, H.B.; Cherif-Silini, H.; Chenari Bouket, A.; Qader, M.; Silini, A.; Yahiaoui, B.; Alenezi, F.N.; Luptakova, L.; Triki, M.A.; Vallat, A.; et al. Screening for Fusarium antagonistic bacteria from contrasting niches designated the endophyte Bacillus halotolerans as plant warden against Fusarium. Front. Microbiol 2019, 9, 3236. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tan, M.; Ma, J.; Zhang, S.; Li, G.; Qu, J. Efficient remediation of PAH-metal co-contaminated soil using microbial-plant combination: A greenhouse study. J. Hazard. Mater. 2016, 302, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Syranidou, E.; Christofilopoulos, S.; Politi, M.; Weyens, N.; Venieri, D.; Vangronsveld, J.; Kalogerakis, N. Bisphenol-A removal by the halophyte Juncus acutus in a phytoremediation pilot: Characterization and potential role of the endophytic community. J. Hazard. Mater. 2017, 323, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xiong, Z.; Chu, L.; Li, W.; Soares, M.A.; White, J.F.; Li, H. Bacterial communities of three plant species from Pb-Zn contaminated sites and plant-growth promotional benefits of endophytic Microbacterium sp. (strain BXGe71). J. Hazard. Mater. 2019, 370, 225–231. [Google Scholar] [CrossRef]
- Pramanik, K.; Mitra, S.; Sarkar, A.; Maiti, T.K. Alleviation of phytotoxic effects of cadmium on rice seedlings by cadmium resistant PGPR strain Enterobacter aerogenes MCC 3092. J. Hazard. Mater. 2018, 351, 317–329. [Google Scholar] [CrossRef]
- Yergeau, E.; Tremblay, J.; Joly, S.; Labrecque, M.; Maynard, C.; Pitre, F.E.; St-Arnaud, M.; Greer, C.W. Soil contamination alters the willow root and rhizosphere metatranscriptome and the root–rhizosphere interactome. ISME J. 2018, 12, 869–884. [Google Scholar] [CrossRef]
- Lumactud, R.; Fulthorpe, R.R. Endophytic bacterial community structure and function of herbaceous plants from petroleum hydrocarbon contaminated and non-contaminated sites. Front. Microbiol. 2018, 9, 1926. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Jin, D.; Jin, S.; Wang, Z.; Yin, H.; Xu, M.; Deng, Y. Responses of bacterial community to dibutyl phthalate pollution in a soil-vegetable ecosystem. J. Hazard. Mater. 2018, 353, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, H.; Dong, Y.; He, Y.; Liu, C. Isolation of vanadium-resistance endophytic bacterium PRE01 from Pteris vittata in stone coal smelting district and characterization for potential use in phytoremediation. J. Hazard. Mater. 2018, 341, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Neetha, J.N.; Sandesh, K.; Girish Kumar, K.; Chidananda, P.; Ujwal, P.C. Optimization of direct blue-14 dye degradation by Bacillus fermus (KX898362) an alkaliphilic plant endophyte and assessment of degraded metabolite toxicity. J. Hazard. Mater. 2019, 364, 742–751. [Google Scholar] [CrossRef]
- Rekik, I.; Chaabane, Z.; Missaoui, A.; Chenari Bouket, A.; Luptakova, L.; Elleuch, A.; Belbahri, L. Effects of untreated and treated wastewater at the morphological, physiological and biochemical levels on seed germination and development of sorghum (Sorghum bicolor (L.) Moench), alfalfa (Medicago sativa L.) and fescue (Festuca arundinacea Schreb.). J. Hazard. Mater. 2017, 326, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Rijavec, T.; Lapanje, A. Hydrogen cyanide in the rhizosphere: Not suppressing plant pathogens, but rather regulating availability of phosphate. Front. Microbiol. 2016, 7, 1785. [Google Scholar] [CrossRef] [PubMed]
- Aloo, B.N.; Makumba, B.A.; Mbega, E.R. The potential of bacilli rhizobacteria for sustainable crop production and environmental sustainability. Microbiol. Res. 2019, 219, 26–39. [Google Scholar] [CrossRef]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.-É.; Carmack, E.C.; Lovejoy, C. Arctic ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.T.; Ung, P.M.U.; Zajkowski, J.; Garneau-Tsodikova, S.; Sherman, D.H. Automated genome mining for natural products. BMC Bioinf. 2009, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; de Jong, A.; Montalbán-López, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal post translationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Ozer, E.A.; Allen, J.P.; Hauser, A.R. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genom. 2014, 15, 737. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Rashad, A.M. Phosphogypsum as a construction material. J. Clean. Prod. 2017, 166, 732–743. [Google Scholar] [CrossRef]
- Marasco, R.; Mosqueira, M.J.; Fusi, M.; Ramond, J.-B.; Merlino, G.; Booth, J.M.; Maggs-Kölling, G.; Cowan, D.A.; Daffonchio, D. Rhizosheath microbial community assembly of sympatric desert speargrasses is independent of the plant host. Microbiome 2018, 6, 215. [Google Scholar] [CrossRef]
- Gonzalez, E.; Pitre, F.E.; Pagé, A.P.; Marleau, J.; Nissim, W.G.; St-Arnaud, M.; Labrecque, M.; Joly, S.; Yergeau, E.; Brereton, N.J.B. Trees, fungi and bacteria: Tripartite metatranscriptomics of a root microbiome responding to soil contamination. Microbiome 2018, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.A.; Durán, P.; Hacquard, S. 2018. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Cernava, T.; Erlacher, A.; Soh, J.; Sensen, C.W.; Grube, M.; Berg, G. Enterobacteriaceae dominate the core microbiome and contribute to the resistome of arugula (Eruca sativa Mill.). Microbiome 2019, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Abdelfattah, A.; Norelli, J.; Burchard, E.; Schena, L.; Droby, S.; Wisniewski, M. Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype-specific influence. Microbiome 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Hartman, K.; van der Heijden, M.G.A.; Roussely-Provent, V.; Walser, J.-C.; Klaus Schlaeppi, K. Deciphering composition and function of the root microbiome of a legume plant. Microbiome 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Kolekar, P.D.; Patil, S.M.; Suryavanshi, M.V.; Suryawanshi, S.S.; Khandare, R.V.; Govindwar, S.P.; Jadhav, J.P. Microcosm study of atrazine bioremediation by indigenous microorganisms and cytotoxicity of biodegraded metabolites. J. Hazard. Mater. 2019, 374, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Roy, A.; Pal, S.; Mohapatra, B.; Kazy, S.K.; Maitia, M.K.; Sar, P. Enrichment and characterization of hydrocarbon-degrading bacteria from petroleum refinery waste as potent bioaugmentation agent for in situ bioremediation. Bioresour. Technol. 2017, 242, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, X.; Gong, J.; Liu, L.; Yi, Y. Specialized core bacteria associate with plants adapted to adverse environment with high calcium contents. PLoS ONE 2018, 13, e0194080. [Google Scholar] [CrossRef]
- Zeng, J.; Zhu, Q.; Li, Y.; Dai, Y.; Wu, Y.; Sun, Y.; Miu, L.; Chen, H.; Lin, X. Isolation of diverse pyrene-degrading bacteria via introducing readily utilized phenanthrene. Chemosphere 2019, 222, 534–540. [Google Scholar] [CrossRef]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Liu, Y.; Du, J.; Lai, Q.; Zeng, R.; Ye, D.; Xu, J.; Shao, Z. Proposal of nine novel species of the Bacillus cereus group. Int. J. Syst. Evol. Microbiol. 2017, 67, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Seghir Daas, M.; Rosana, A.R.R.; Acedo, J.Z.; Douzane, M.; Nateche, F.; Kebbouche-Gana, S.; Vederas, J.C. Insights into the draft genome sequence of bioactives-producing Bacillus thuringiensis DNG9 isolated from Algerian soil-oil slough. Stand. Genom. Sci. 2018, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D.; et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 4894. [Google Scholar] [CrossRef] [PubMed]











 Tunisia,
Tunisia,  Pacific of Ocean,
Pacific of Ocean,  the United States,
the United States,  China,
China,  France,
France,  Japan,
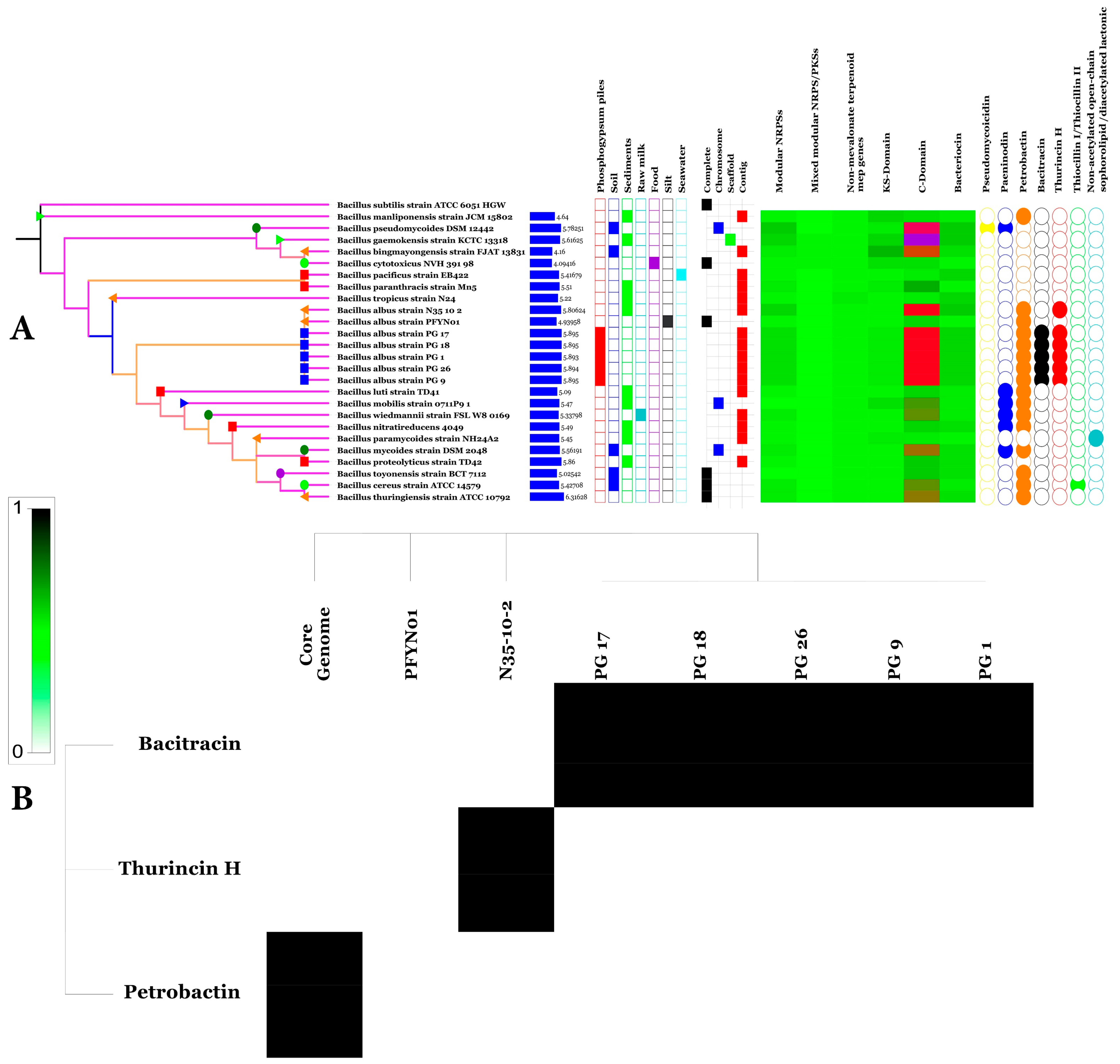
Japan,  South Korea) (A), genome size, isolation source, level of sequencing, prevalence of secondary metabolite clusters and types genes (from left to right), Heat map of secondary metabolite prevalence among BA core and accessory genomes (B).
Tunisia, Pacific of Ocean, the United States, China, France, Japan, South Korea) (A), genome size, isolation source, level of sequencing, prevalence of secondary metabolite clusters and types genes (from left to right), Heat map of secondary metabolite prevalence among BA core and accessory genomes (B).
South Korea) (A), genome size, isolation source, level of sequencing, prevalence of secondary metabolite clusters and types genes (from left to right), Heat map of secondary metabolite prevalence among BA core and accessory genomes (B).
Tunisia, Pacific of Ocean, the United States, China, France, Japan, South Korea) (A), genome size, isolation source, level of sequencing, prevalence of secondary metabolite clusters and types genes (from left to right), Heat map of secondary metabolite prevalence among BA core and accessory genomes (B).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Mefteh, F.; Chenari Bouket, A.; Daoud, A.; Luptakova, L.; N. Alenezi, F.; Gharsallah, N.; Belbahri, L. Metagenomic Insights and Genomic Analysis of Phosphogypsum and Its Associated Plant Endophytic Microbiomes Reveals Valuable Actors for Waste Bioremediation. Microorganisms 2019, 7, 382. https://doi.org/10.3390/microorganisms7100382
Ben Mefteh F, Chenari Bouket A, Daoud A, Luptakova L, N. Alenezi F, Gharsallah N, Belbahri L. Metagenomic Insights and Genomic Analysis of Phosphogypsum and Its Associated Plant Endophytic Microbiomes Reveals Valuable Actors for Waste Bioremediation. Microorganisms. 2019; 7(10):382. https://doi.org/10.3390/microorganisms7100382
Chicago/Turabian StyleBen Mefteh, Fedia, Ali Chenari Bouket, Amal Daoud, Lenka Luptakova, Faizah N. Alenezi, Neji Gharsallah, and Lassaad Belbahri. 2019. "Metagenomic Insights and Genomic Analysis of Phosphogypsum and Its Associated Plant Endophytic Microbiomes Reveals Valuable Actors for Waste Bioremediation" Microorganisms 7, no. 10: 382. https://doi.org/10.3390/microorganisms7100382
APA StyleBen Mefteh, F., Chenari Bouket, A., Daoud, A., Luptakova, L., N. Alenezi, F., Gharsallah, N., & Belbahri, L. (2019). Metagenomic Insights and Genomic Analysis of Phosphogypsum and Its Associated Plant Endophytic Microbiomes Reveals Valuable Actors for Waste Bioremediation. Microorganisms, 7(10), 382. https://doi.org/10.3390/microorganisms7100382