Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease—Is There Cause for Consideration?




{kind=link}
{kind=link}
Abstract
:1. Introduction
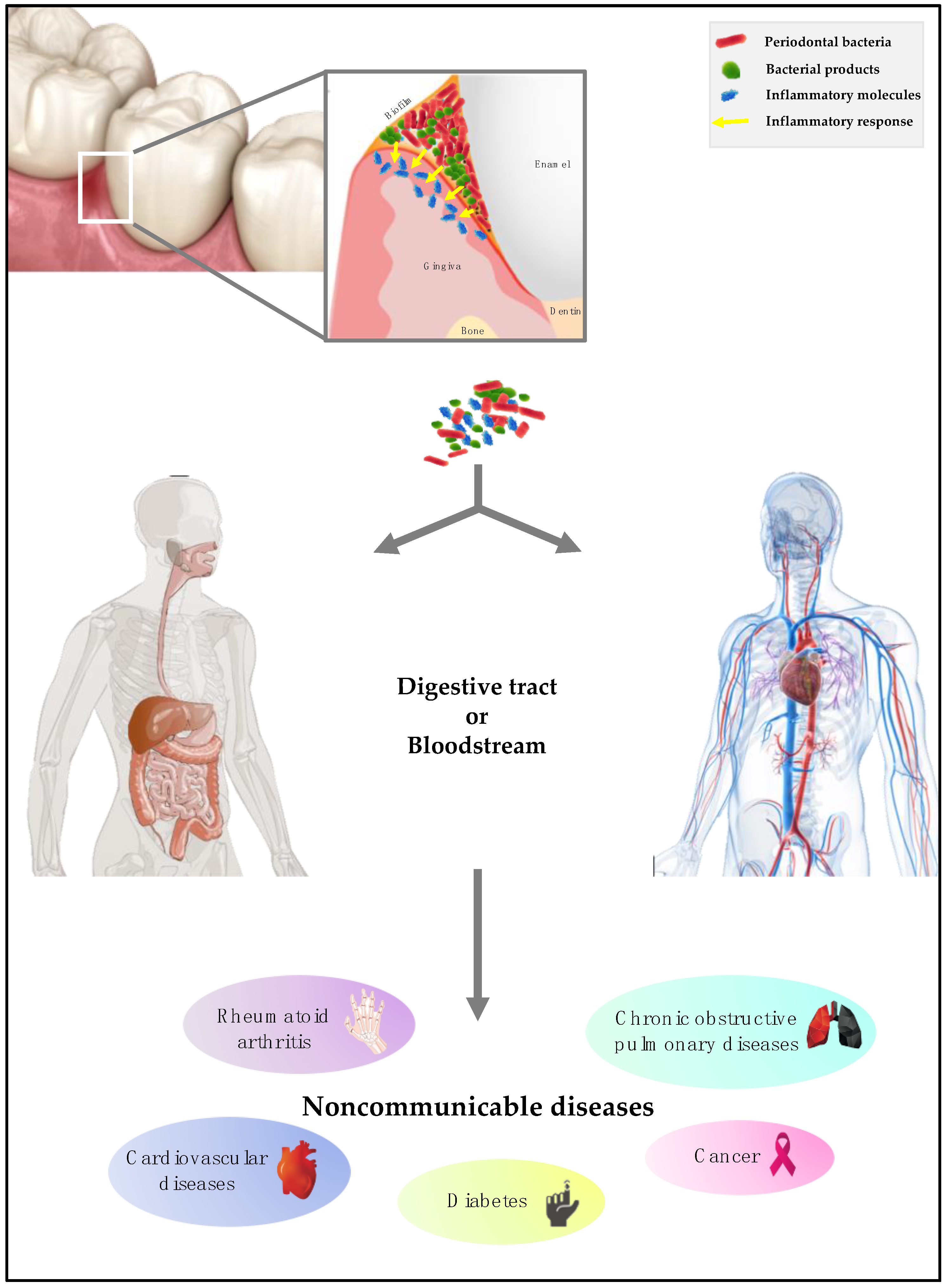
2. Periodontal Disease
3. The Invasion Process by Periodontal Bacteria
4. Periodontal Pathogens and Diabetes
5. Periodontal Pathogens and Cardiovascular Diseases
6. Periodontal Pathogens and Chronic Obstructive Pulmonary Disease
7. Periodontal Pathogens and Rheumatoid Arthritis
8. Periodontal Pathogens and Cancer
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Petersen, P.E. World Health Organization global policy for improvement of oral health-World Health Assembly 2007. Int. Dent. J. 2008, 58, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.E. Global policy for improvement of oral health in the 21st century–Implications to oral health research of World Health Assembly 2007, World Health Organization. Commun. Dent. Oral Epidemiol. 2009, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- World Health Assembly, 69. Prevention and Control of Noncommunicable Diseases: Responses to Specific Assignments in Preparation for the Third High-Level Meeting of the United Nations General Assembly on the Prevention and Control of Non-Communicable Diseases in 2018: Report by the Director-General; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- FDI World Dental Federation. World Health Assembly, 69–Prevention and Control of Noncommunicable Diseases. Agenda Item: 12.4: Prevention and Control of Noncommunicable Diseases: Responses to Specific Assignments in Preparation for the Third High-Level Meeting of the United Nations General Assembly on the Prevention and Control of Noncommunicable Diseases in 2018. Available online: https://www.fdiworlddental.org/resources/world-health-assembly-statements/wha69-prevention-and-control-of-noncommunicable-diseases (accessed on 29 September 2019).
- Kassier, S.M. Periodontal disease and non-communicable diseases. Strength of bidirectional associations. South Afr. Dent. J. 2016, 71, 404–409. [Google Scholar] [CrossRef]
- Petersen, P.E.; Bourgeois, D.; Ogawa, H.; Estupinan-Day, S.; Ndiaye, C. The global burden of oral diseases and risks to oral health. Bull. World Health Organ. 2005, 83, 661–669. [Google Scholar] [PubMed]
- Türp, J.C.; Spranger, H. Non-communicable disease and their significance for dental medicine. Swiss Dent. J. 2016, 126, 473–489. [Google Scholar] [PubMed]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2006, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Arweiler, N.B.; Netuschlil, L. The Oral Microbiota. Adv. Exp. Med. Biol. 2016, 902, 45–60. [Google Scholar] [CrossRef]
- Sampaio-Maia, B.; Caldas, I.M.; Pereira, M.L.; Pérez-Mongiovi, D.; Araujo, R. The Oral Microbiome in Health and Its Implication in Oral and Systemic Diseases. Adv. Appl. Microbiol. 2016, 97, 171–210. [Google Scholar] [CrossRef]
- Gao, L.; Xu, T.; Huang, G.; Jiang, S.; Gu, Y.; Chen, F. Oral microbiomes: More and more importance in oral cavity and whole body. Protein Cell 2018, 9, 488–500. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Li, H.; Ni, C.; Du, Z.; Yan, F. Human oral microbiota and its modulation for oral health. Biomed. Pharmacother. 2018, 99, 883–893. [Google Scholar] [CrossRef]
- Bartold, P.M.; Van Dyke, T.E. Periodontitis: A host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontology 2013, 62, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Costalonga, M.; Herzberg, M.C. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol. Lett. 2014, 162, 22–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- AlJehani, Y.A. Risk factors of periodontal disease: Review of the literature. Int. J. Dent. 2014, 2014, 182513. [Google Scholar] [CrossRef] [PubMed]
- Linden, G.J.; Hersberg, M.C.; Working group 4 of the joint EFP/AAP workshop. Periodontitis and systemic diseases: A record of discussions of working group 4 of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Periodontol. 2013, 84, S20–S23. [Google Scholar] [CrossRef]
- Holmstrup, P.; Damgaard, C.; Olsen, I.; Klinge, B.; Flyvbjerg, A.; Nielsen, C.H.; Hansen, P.R. Comorbidity of periodontal disease: Two sides of the same coin? An introduction for the clinician. J. Oral Microbiol. 2017, 9, 1332710. [Google Scholar] [CrossRef] [PubMed]
- Nwhator, S.O.; Heikkinen, A.M.; Tervahartiala, T.; Gieselmann, D.R.; Leppilahti, J.; Sorsa, T. aMMP-8 Oral Fluid PoC Test. In Translational Oral Health Research; Meurman, J.H., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 33–41. [Google Scholar]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Carrouel, F.; Viennot, S.; Santamaria, J.; Veber, P.; Bourgeois, D. Quantitative Molecular Detection of 19 Major Pathogens in the Interdental Biofilm of Periodontally Healthy Young Adults. Front. Microbiol. 2016, 7, 840. [Google Scholar] [CrossRef]
- Suzuki, N.; Yoneda, M.; Hirofuji, T. Mixed red-complex bacterial infection in periodontitis. Int. J. Dent. 2013, 2013, 587279. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef]
- Rosier, B.T.; De Jager, M.; Zaura, E.; Krom, B.P. Historical and contemporary hypotheses on the development of oral diseases:are we there yet? Front. Cell Infect. Microbiol. 2014, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Tribble, G.D.; Baker, H.V.; Mans, J.J.; Handfield, M.; Lamont, R.J. Role of Porphyromonas gingivalis SerB in gingival epithelial cell cytoskeletal remodeling and cytokine production. Infect. Immun. 2008, 76, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Belton, C.M.; Reife, R.A.; Lamont, R.J. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect. Immun. 1998, 66, 1660–1665. [Google Scholar] [PubMed]
- Ji, S.; Kim, Y.; Min, B.M.; Han, S.H.; Choi, Y. Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. J. Periodontal Res. 2007, 42, 503–510. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Stover, C.M.; Dupont, A.P. Gingivalis in Periodontal Disease and Atherosclerosis–Scenes of Action for Antimicrobial Peptides and Complement. Front. Immunol. 2015, 6, 45. [Google Scholar] [CrossRef]
- Hugues, F. Periodontium and Periodontal Disease. In Stem Cell Biology and Tissue Engineering in Dental Sciences, 1st ed.; Vishwakarma, A., Sharpe, P., Shi, S., Ramalingam, M., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 433–444. [Google Scholar]
- Wang, G.P. Defining functional signatures of dysbiosis in periodontitis progression. Genome Med. 2015, 7, 40. [Google Scholar] [CrossRef]
- Vieira Colombo, A.P.; Magalhães, C.B.; Hartenbach, F.A.R.R.; Martins do Souto, R.; Maciel da Silva-Boghossian, C. Periodontal-disease-associated biofilm: A reservoir for pathogens of medical importance. Microb. Pathog. 2016, 94, 27–34. [Google Scholar] [CrossRef]
- Popova, C.; Dosseva-Panova, V.; Panov, V. Microbiology of Periodontal Diseases. Biotechnol. Biotechnol. Equip. 2013, 27, 3754–3759. [Google Scholar] [CrossRef]
- Li, X.; Kolltveit, K.M.; Tronstad, L.; Olsen, I. Systemic diseases caused by oral infection. Clin. Microb. Rev. 2000, 13, 547–558. [Google Scholar] [CrossRef]
- Friedewald, V.E.; Komman, K.S.; Beck, J.D.; Genco, R.; Goldfine, A.; Libby, P.; Offenbacher, S.; Ridker, P.M.; Van Dike, T.E.; Roberts, W.C. The American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: Periodontitis and atherosclerotic cardiovascular disease. Am. J. Cardiol. 2009, 104, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Q.; Chen, M.; Wu, W.Z.; Wang, R.; Liu, C.J.; Li, B.; Shi, X.L.; Du, H.S.; Tan, H.B. Association Between Helicobacter pylori Infection and Risk of Periodontal Diseases in Han Chinese: A Case-Control Study. Med. Sci. Monit. 2016, 22, 121–126. [Google Scholar] [CrossRef]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H.; et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huuttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Dian, D.; Keshavarzian, A.; Fogg, L.; Fields, J.Z.; Farhadi, A. The Role of Oral Hygiene in Inflammatory Bowel Disease. Dig. Dis. Sci. 2011, 56, 170–175. [Google Scholar] [CrossRef]
- Flynn, K.J.; Baxter, N.T.; Schloss, P.D. Metabolic and Community Synergy of Oral Bacteria in Colorectal Cancer. Msphere 2016, 1, e00102-16. [Google Scholar] [CrossRef]
- Casanova, L.; Hughes, F.J.; Preshaw, P.M. Diabetes and periodontal disease: A two-way relationship. Br. Dent. J. 2014, 217, 433–437. [Google Scholar] [CrossRef]
- Branchereau, M.; Reichardt, F.; Loubieres, P.; Marck, P.; Waget, A.; Azalbert, V.; Colom, A.; Padmanabhan, R.; Iacovoni, J.S.; Giry, A.; et al. Periodontal dysbiosis linked to periodontitis is associated with car-diometabolic adaptation to high-fat diet in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1091–G1101. [Google Scholar] [CrossRef]
- Burcelin, R.; Serino, M.; Chabo, C.; Blasco-Baque, V.; Amar, J. Gut microbiota and diabetes: From pathogenesis to therapeutic perspective. Acta Diabetol. 2011, 48, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Tunes, R.S.; Foss-Freitas, M.C.; Nogueira-Filho, G.D.R. Impact of periodontitis on the diabetes-related inflammatory status. J. Can. Dent. Assoc. 2010, 76, a35. [Google Scholar]
- Blasco-Baque, V.; Garidou, L.; Pomie, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef]
- Liu, L.S.; Gkranias, N.; Farias, B.; Spratt, D.; Donos, N. Differences in the subgingival microbial population of chronic periodontitis in subjects with and without type 2 diabetes mellitus-a systematic review. Clin. Oral. Investig. 2018, 22, 2743–2762. [Google Scholar] [CrossRef]
- Saffi, M.A.; Furtado, M.V.; Polanczyk, C.A.; Montenegro, M.M.; Ribeiro, I.W.; Kampits, C.; Haas, A.N.; Rösing, C.K.; Rabelo-Silva, E.R. Relationship between vascular endothelium and periodontal disease in atherosclerotic lesions: Review article. World J. Cardiol. 2015, 7, 26–30. [Google Scholar] [CrossRef]
- Gubern, C.; López-Bermejo, A.; Biarnés, J.; Vendrell, J.; Ricart, W.; Fernández-Real, J.M. Natural antibiotics and insulin sensitivity: The role of bactericidal/permeability-increasing protein. Diabetes 2006, 55, 216–224. [Google Scholar] [CrossRef]
- Vrieze, A.; Holleman, F.; Zoetendal, E.G.; de Vos, W.M.; Hoekstra, J.B.; Nieuwdorp, M. The environment within: How gut microbiota may influence metabolism and body composition. Diabetologia 2010, 53, 606–613. [Google Scholar] [CrossRef]
- Batista, R.M.; Rosetti, E.P.; Zandonade, E.; Roelke, L.H.; Vettore, M.V.; Oliveira, A.E. Association between periodontal disease and subclinical atherosclerosis: The ELSA-Brasil study. Cad. Saude Publica 2012, 28, 965–976. [Google Scholar] [CrossRef]
- Stewart, R.; West, M. Increasing evidence for an association between periodontitis and cardiovascular disease. Circulation 2016, 133, 549–551. [Google Scholar] [CrossRef]
- Aarabi, G.; Heydecke, G.; Seedorf, U. Roles of Oral Infections in the Pathomechanism of Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1978. [Google Scholar] [CrossRef]
- Chun, Y.H.; Chun, K.R.; Olguin, D.; Wang, H.L. Biological foundation for periodontitis as a potential risk factor for atherosclerosis. J. Periodont. Res. 2005, 40, 87–95. [Google Scholar] [CrossRef]
- Khlgatian, M.; Nassar, H.; Chou, H.H.; Gibson, F.C.; Genco, C.A. Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells. Infect. Immun. 2002, 70, 257–267. [Google Scholar] [CrossRef]
- Brinson, C.W.; Lu, Z.; Li, Y.; Lopes-Virella, M.F.; Huang, Y. Lipopolysaccharide and IL-1β coordinate a synergy on cytokine production by upregulating MyD88 expression in human gingival fibroblasts. Mol. Immunol. 2016, 79, 47–54. [Google Scholar] [CrossRef]
- Fernandes, C.P.; Oliveira, F.A.; Silva, P.G.; Alves, A.P.; Mota, M.R.; Montenegro, R.C.; Burbano, R.M.; Seabra, A.D.; Lobo Filho, J.G.; Lima, D.L.; et al. Molecular analysis of oral bacteria in dental biofilm and atherosclerotic plaques of patients with vascular disease. Int. J. Cardiol. 2014, 174, 710–712. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, C.; Viereck, J.; Hua, N.; Phinikaridou, A.; Madrigal, A.G.; Gibson, F.C.; Hamilton, J.A.; Genco, C.A. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 2011, 215, 52–59. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Alfthan, G.; Tuomilehto, J.; Asikainen, S.; Jousilahti, P. High serum antibody levels to Porphyromonas gingivalis predict myocardial infarction. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 408–411. [Google Scholar] [CrossRef]
- Xu, S.; Song, M.; Xiong, Y.; Liu, X.; He, Y.; Qin, Z. The association between periodontal disease and the risk of myocardial infarction: A pooled analysis of observational studies. BMC Cardiovasc. Disord. 2017, 17, 50. [Google Scholar] [CrossRef]
- Ohki, T.; Itabashi, Y.; Kohno, T.; Yoshizawa, A.; Nishikubo, S.; Watanabe, S.; Yamane, G.; Ishihara, K. Detection of periodontal bacteria in thrombi of patients with acute myocardial infarction by polymerase chain reaction. Am. Heart J. 2012, 163, 164–167. [Google Scholar] [CrossRef]
- Lund Håheim, L.; Olsen, I.; Nafstad, P.; Schwarze, P.; Rønningen, K.S. Antibody levels to single bacteria or in combination evaluated against myocardial infarction. J. Clin. Periodontol. 2008, 35, 473–478. [Google Scholar] [CrossRef]
- Pessi, T.; Karhunen, V.; Karjalainen, P.P.; Ylitalo, A.; Airaksinen, J.K.; Niemi, M.; Pietila, M.; Lounatmaa, K.; Haapaniemi, T.; Lehtimäki, T.; et al. Bacterial Signatures in Thrombus Aspirates of Patients with Myocardial Infarction Clinical Perspective. Circulation 2013, 127, 1219–1228. [Google Scholar] [CrossRef]
- Ammirati, E.; Maseri, A. Letter by Ammirati and Maseri Regarding Article, “Bacterial Signatures in Thrombus Aspirates of Patients with Myocardial Infarction”. Circulation 2013, 128, e235. [Google Scholar] [CrossRef]
- Pessi, T.; Karhunen, V.; Karjalainen, P.P.; Ylitalo, A.; Airaksinen, J.K.; Niemi, M.; Pietila, M.; Lounatmaa, K.; Haapaniemi, T.; Lehtimäki, T.; et al. Response to Letters Regarding Article, “Bacterial Signatures in Thrombus Aspirates of Patients with Myocardial Infarction”. Circulation 2013, 128, e237–e238. [Google Scholar] [CrossRef]
- Srisuwantha, R.; Shiheido, Y.; Aoyama, N.; Sato, H.; Kure, K.; Laosrisin, N.; Izumi, Y.; Suzuki, J.I. Porphyromonas Gingivalis Elevated High-Mobility Group Box 1 Levels After Myocardial Infarction in Mice. Int. Heart J. 2017, 58, 762–768. [Google Scholar] [CrossRef]
- Shiheido, Y.; Maejima, Y.; Suzuki, J.I.; Aoyama, N.; Kaneko, M.; Watanabe, R.; Sakamaki, Y.; Wakayama, K.; Ikeda, Y.; Akazawa, H.; et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J. Mol. Cell. Cardiol. 2016, 99, 123–137. [Google Scholar] [CrossRef]
- Suzuki, J.I.; Sato, H.; Kaneko, M.; Yoshida, A.; Aoyama, N.; Akimoto, S.; Wakayama, K.; Kumagai, H.; Ikeda, Y.; Akazawa, H.; et al. Periodontitis and myocardial hypertrophy. Hypertens. Res. 2017, 40, 324–328. [Google Scholar] [CrossRef]
- Lee, Y.L.; Hu, H.Y.; Chou, P.; Chu, D. Dental prophylaxis decreases the risk of acute myocardial infarction: A nationwide population-based study in Taiwan. Clin. Interv. Aging 2015, 10, 175–182. [Google Scholar] [CrossRef]
- Lafon, A.; Pereira, B.; Dufour, T.; Rigouby, V.; Giroud, M.; Béjot, Y.; Tubert-Jeannin, S. Periodontal disease and stroke: A meta-analysis of cohort studies. Eur. J. Neurol. 2014, 21, 1155–1161. [Google Scholar] [CrossRef]
- Sen, S.; Giamberardino, L.D.; Moss, K.; Morelli, T.; Rosamond, W.D.; Gottesman, R.F.; Beck, J.; Offenbacher, S. Periodontal Disease, Regular Dental Care Use, and Incident Ischemic Stroke. Stroke 2018, 49, 355–362. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Alfthan, G.; Jousilahti, P. Systemic exposure to Pophyromonas gingivalis predicts incident stroke. Atherosclrosis 2007, 193, 222–228. [Google Scholar] [CrossRef]
- Funaki, S.; Tokutomi, F.; Wada-Takahashi, S.; Yoshino, F.; Yoshida, A.; Maehata, Y.; Miyamoto, C.; Toyama, T.; Sato, T.; Hamada, N.; et al. Porphyromonas gingivalis infection modifies oral microcirculation and aortic vascular function in the stroke-prone spontaneously hypertensive rat (SHRSP). Microb. Pathog. 2016, 92, 36–42. [Google Scholar] [CrossRef]
- Meurman, J.H. A Role for Oral Health Personnel in Stroke Prevention. Compend. Contin. Educ. Dent. 2017, 38, 26–28. [Google Scholar]
- Söder, B.; Meurman, J.H.; Söder, P.Ö. Gingival Inflammation Associates with Stroke-A Role for Oral Health Personnel in Prevention: A Database Study. PLoS ONE 2015, 10, e0137142. [Google Scholar] [CrossRef]
- Revest, M.; Egmann, G.; Cattoir, V.; Tattevin, P. HACEK endocarditis: State-of-the-art. Expert Rev. Anti Infect. Ther. 2016, 14, 523–530. [Google Scholar] [CrossRef]
- Lindholm, M.; Min Aung, K.; Nyunt Wai, S.; Oscarsson, J. Role of OmpA1 and OmpA2 in Aggregatibacter actinomycetemcomitans and Aggregatibacter aphrophilus serum resistance. J. Oral. Microbiol. 2018, 11, 1536192. [Google Scholar] [CrossRef]
- Brown, A.C.; Boesze-Battaglia, K.; Balashova, N.V.; Mas Gómez, N.; Speicher, K.; Tang, H.Y.; Duszyk, M.E.; Lally, E.T. Membrane localization of the Repeats-in-Toxin (RTX) Leukotoxin (LtxA) produced by Aggregatibacter actinomycetemcomitans. PLoS ONE 2018, 13, e0205871. [Google Scholar] [CrossRef]
- Zhu, B.; Macleod, L.C.; Kitten, T.; Xu, P. Streptococcus sanguinis biofilm formation & interaction with oral pathogens. Future Microbiol. 2018, 13, 915–932. [Google Scholar] [CrossRef]
- Cahill, T.J.; Prendergast, B.D. Infective endocarditis. Lancet 2016, 387, 882–893. [Google Scholar] [CrossRef]
- Moser, C.; Pedersen, H.T.; Lerche, C.J.; Kolpen, M.; Line, L.; Thomsen, K.; Høiby, N.; Jensen, P.Ø. Biofilms and host response–Helpful or harmful. APMIS 2017, 125, 320–338. [Google Scholar] [CrossRef]
- Forum of International Respiratory Societies; European Respiratory Society. The Global Impact of Respiratory Disease; Forum of International Respiratory Societies: Lausanne, Switzerland, 2017; ISBN 978-1-84984-087-3. [Google Scholar]
- GBD. Compare|IHME Viz Hub. Available online: http://vizhub.healthdata.org/gbd-compare (accessed on 29 September 2019).
- Andreas, S.; Hering, T.; Mühlig, S.; Nowak, D.; Raupach, T.; Worth, H. Smoking cessation in chronic obstructive pulmonary disease: An effective medical intervention. Dtsch. Arzteblatt Int. 2009, 106, 276–282. [Google Scholar]
- Usher, A.K.; Stockley, R.A. The link between chronic periodontitis and COPD: A common role for the neutrophil? BMC Med. 2013, 11, 241. [Google Scholar] [CrossRef]
- Hobbins, S.; Chapple, I.L.; Sapey, E.; Stockley, R.A. Is periodontitis a comorbidity of COPD or can associations be explained by shared risk factors/behaviors? Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 1339–1349. [Google Scholar] [CrossRef]
- Muthu, J.; Muthanandam, S.; Mahendra, J. Mouth the mirror of lungs: Where does the connection lie? J. Front. Med. 2016, 10, 405–409. [Google Scholar] [CrossRef]
- Page, R.C.; Eke, P.I. Case definitions for use in population-based surveillance of periodontitis. J. Periodontol. 2007, 78, 1387–1399. [Google Scholar] [CrossRef]
- Hasegawa, A.; Sato, T.; Hoshikawa, Y.; Ishida, N.; Tanda, N.; Kawamura, Y.; Kondo, T.; Takahashi, N. Detection and identification of oral anaerobes in intraoperative bronchial fluids of patients with pulmonary carcinoma. Microbiol. Immunol. 2014, 58, 375–381. [Google Scholar] [CrossRef]
- Han, M.K.; Huang, Y.J.; Lipuma, J.J.; Boushey, H.A.; Boucher, R.C.; Cookson, W.O.; Curtis, J.L.; Erb-Downward, J.; Lynch, S.V.; Sethi, S.; et al. Significance of the microbiome in obstructive lung disease. Thorax 2012, 67, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Zhang, B.; Xing, H.; Yang, S.; Xu, J.; Liu, H. Patients with Chronic Obstructive Pulmonary Disease Suffer from Worse Periodontal Health-Evidence from a Meta-Analysis. Front. Physiol. 2018, 9, 33. [Google Scholar] [CrossRef]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.; Shu, D.; Xiong, Y.; He, M.; Xu, S.; Guo, B. Association of Periodontitis with Rheumatoid Arthritis and the Effect of Non-Surgical Periodontal Treatment on Disease Activity in Patients with Rheumatoid Arthritis. Med. Sci. Monit. 2018, 24, 5802–5810. [Google Scholar] [CrossRef]
- Araújo, V.M.; Melo, I.M.; Lima, V. Relationship between Periodontitis and Rheumatoid Arthritis: Review of the Literature. Med. Inflamm. 2015, 2015, 259074. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- McHugh, J. Rheumatoid arthritis: New model linking periodontitis and RA. Nat. Rev. Rheumatol. 2017, 13, 66. [Google Scholar] [CrossRef]
- Reichert, S.; Haffner, M.; Keyber, G.; Schäfer, C.; Stein, J.M.; Schaller, H.G.; Wienke, A.; Strauss, H.; Heide, S.; Schulz, S. Detection of oral bacterial DNA in synovial fluid. J. Clin. Periodontol. 2013, 40, 591–598. [Google Scholar] [CrossRef]
- Témoin, S.; Chakaki, A.; Askari, A.; El-Halaby, A.; Fitzgerald, S.; Marcus, R.E.; Han, Y.W.; Bissada, N.F. Identification of oral bacterial DNA in synovial fluid of patients with arthritis with native and failed prosthetic joints. J. Clin. Rheumatol. 2012, 18, 117–121. [Google Scholar] [CrossRef]
- Cheng, Z.; Meade, J.; Mankia, K.; Emery, P.; Devine, D.A. Periodontal disease and periodontal bacteria as triggers for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 19–30. [Google Scholar] [CrossRef]
- Badran, Z.; Struillou, X.; Verner, C.; Clee, T.; Rakic, M.; Martinez, M.C.; Soueidan, A. Periodontitis as a risk factor for systemic disease: Are microparticles the missing link? Med. Hypotheses 2015, 84, 555–556. [Google Scholar] [CrossRef]
- Ribeiro, J.; Leão, A.; Novaes, A.B. Periodontal infection as a possible severity factor for rheumatoid arthritis. J. Clin. Periodontol. 2005, 32, 412–416. [Google Scholar] [CrossRef]
- Nesse, W.; Westra, J.; van der Wal, J.E.; Abbas, F.; Nicholas, A.P.; Vissink, A.; Brouwer, E. The periodontium of periodontitis patients contains citrullinated proteins which may play a role in ACPA (anti-citrullinated protein antibody) formation. J. Clin. Periodontol. 2012, 39, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, A.; Almas, K. Rheumatoid Arthritis and Periodontal Disease. An Update. N.Y. State Dent. J. 2015, 81, 30–36. [Google Scholar]
- Mühlberg, S.; Jäger, J.; Krohn-Grimberghe, B.; Patschan, S.; Mausberg, R.F.; Schmalz, G.; Haak, R.; Ziebolz, D. Oral health-related quality of life depending on oral health in patients with rheumatoid arthritis. Clin. Oral. Investig. 2017, 21, 2661–2670. [Google Scholar] [CrossRef]
- Michaud, D.S.; Fu, Z.; Shi, J.; Chung, M. Periodontal Disease, Tooth Loss, and Cancer Risk. Epidemiol. Rev. 2017, 39, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Moraes, R.C.; Dias, F.L.; Figueredo, C.M.; Fischer, R.G. Association between Chronic Periodontitis and Oral/Oropharyngeal Cancer. Braz. Dent. J. 2016, 27, 261–266. [Google Scholar] [CrossRef]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host. Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef]
- Gao, S.; Li, S.; Ma, Z.; Liang, S.; Shan, T.; Zhang, M.; Zhu, X.; Zhang, P.; Liu, G.; Zhou, F.; et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect. Agents Cancer 2016, 11, 3. [Google Scholar] [CrossRef]
- Nishihara, T.; Koseki, T. Microbial etiology of periodontitis. Periodontology 2004, 36, 14–26. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef]
- Karpiński, T.M. Role of Oral Microbiota in Cancer Development. Microorganisms 2019, 7, 20. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, M.; Salazar, C.R.; Hays, R.; Bedi, S.; Chen, Y.; Li, Y. Chronic Periodontal Disease, Periodontal Pathogen Colonization, and Increased Risk of Precancerous Gastric Lesions. J. Periodontol. 2017, 88, 1124–1134. [Google Scholar] [CrossRef]
- Salazar, C.R.; Sun, J.; Li, Y.; Francois, F.; Corby, P.; Perez-Perez, G.; Dasanayake, A.; Pei, Z.; Chen, Y. Association between selected oral pathogens and gastric precancerous lesions. PLoS ONE 2013, 8, e51604. [Google Scholar] [CrossRef]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef]
- Desai, H.G.; Gill, H.H.; Shankaran, K.; Mehta, P.R.; Prabhu, S.R. Dental plaque: A permanent reservoir of Helicobacter pylori? Scand. J. Gastroenterol. 1991, 26, 1205–1208. [Google Scholar] [CrossRef]
- Anand, P.S.; Kamath, K.P.; Anil, S. Role of dental plaque, saliva and periodontal disease in Helicobacter pylori infection. World J. Gastroenterol. 2014, 20, 5639–5653. [Google Scholar] [CrossRef]
- Song, Q.; Lange, T.; Spahr, A.; Adler, G.; Bode, G. Characteristic distribution pattern of Helicobacter pylori in dental plaque and saliva detected with nested PCR. J. Med. Microbiol. 2000, 49, 349–353. [Google Scholar] [CrossRef]
- Herrera, V.; Parsonnet, J. Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 2009, 15, 971–976. [Google Scholar] [CrossRef]
- Moss, S.F. The Clinical Evidence Linking Helicobacter pylori to Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2016, 3, 183–191. [Google Scholar] [CrossRef]
- Ishihara, K.; Miura, T.; Kimizuka, R.; Ebihara, Y.; Mizuno, Y.; Okuda, K. Oral bacteria inhibit Helicobacter pylori growth. FEMS Microbiol. Lett. 1997, 152, 355–361. [Google Scholar] [CrossRef]
- Umeda, M.; Kobayashi, H.; Takeuchi, Y.; Hayashi, J.; Morotome-Hayashi, Y.; Yano, K.; Aoki, A.; Ohkusa, T.; Ishikawa, I. High prevalence of Helicobacter pylori detected by PCR in the oral cavities of periodontitis patients. J. Periodontol. 2003, 74, 129–134. [Google Scholar] [CrossRef]
- Zou, Q.H.; Li, R.Q. Helicobacter pylori in the oral cavity and gastric mucosa: A meta-analysis. J. Oral Pathol. Med. 2011, 40, 317–324. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourgeois, D.; Inquimbert, C.; Ottolenghi, L.; Carrouel, F. Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease—Is There Cause for Consideration? Microorganisms 2019, 7, 424. https://doi.org/10.3390/microorganisms7100424
Bourgeois D, Inquimbert C, Ottolenghi L, Carrouel F. Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease—Is There Cause for Consideration? Microorganisms. 2019; 7(10):424. https://doi.org/10.3390/microorganisms7100424
Chicago/Turabian StyleBourgeois, Denis, Camille Inquimbert, Livia Ottolenghi, and Florence Carrouel. 2019. "Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease—Is There Cause for Consideration?" Microorganisms 7, no. 10: 424. https://doi.org/10.3390/microorganisms7100424
APA StyleBourgeois, D., Inquimbert, C., Ottolenghi, L., & Carrouel, F. (2019). Periodontal Pathogens as Risk Factors of Cardiovascular Diseases, Diabetes, Rheumatoid Arthritis, Cancer, and Chronic Obstructive Pulmonary Disease—Is There Cause for Consideration? Microorganisms, 7(10), 424. https://doi.org/10.3390/microorganisms7100424