Staphylococcus arlettae Genomics: Novel Insights on Candidate Antibiotic Resistance and Virulence Genes in an Emerging Opportunistic Pathogen

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Sequencing of SAR Bari
2.2. Genome Assembly and Taxonomic Classification of SAR Bari
2.3. Antimicrobial Susceptibility Testing
2.4. Comparative Genomics Dataset
2.5. Genomics of Antibiotic Resistance
2.6. Genomics of Virulence
3. Results
3.1. Genomics and Taxonomy of SAR (Staphylococcus arlettae) Bari
3.2. Antibiotic Resistance of SAR Bari
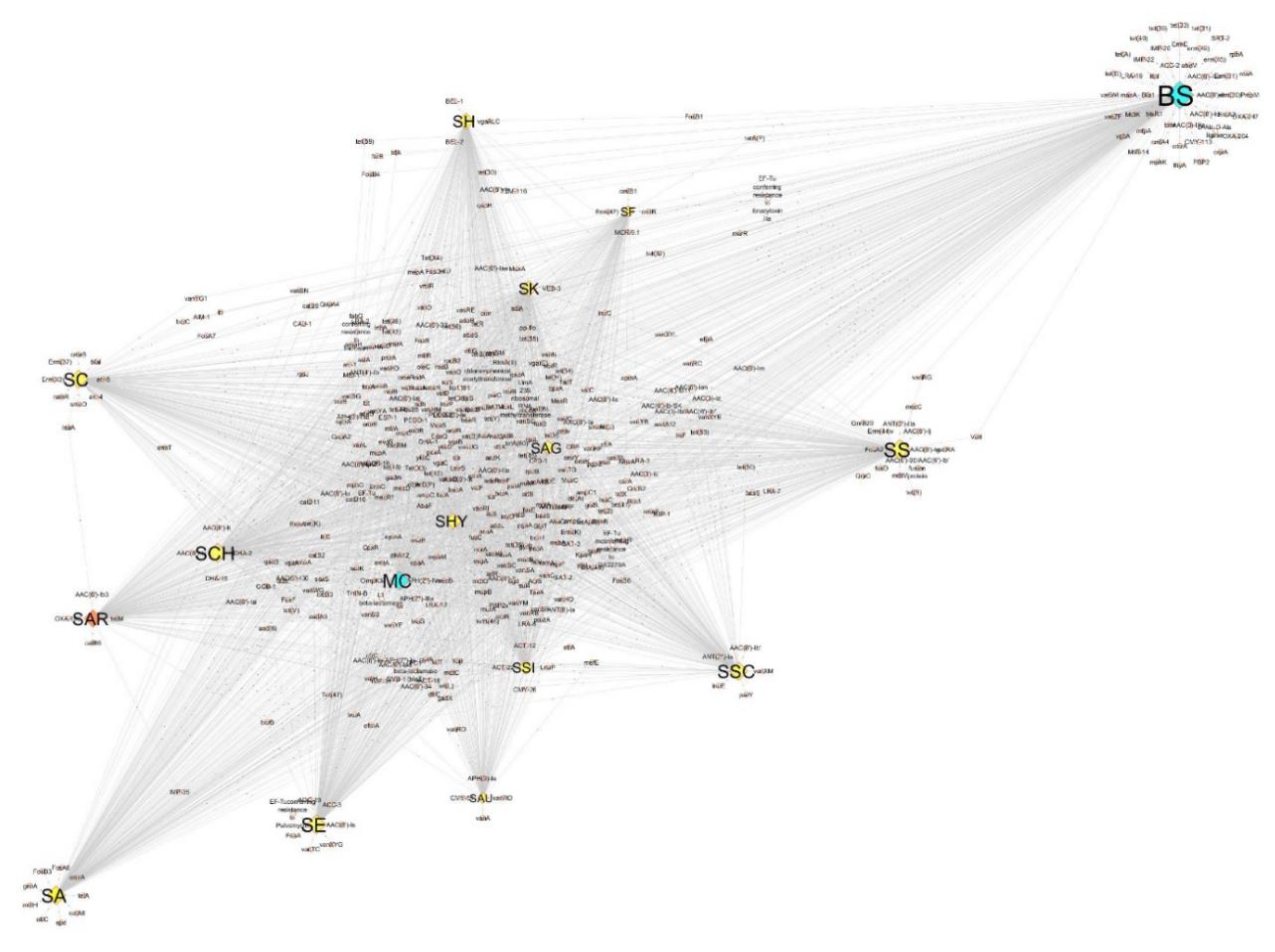
3.3. Antibiotic Resistance Genomics of SAR and Species Dataset
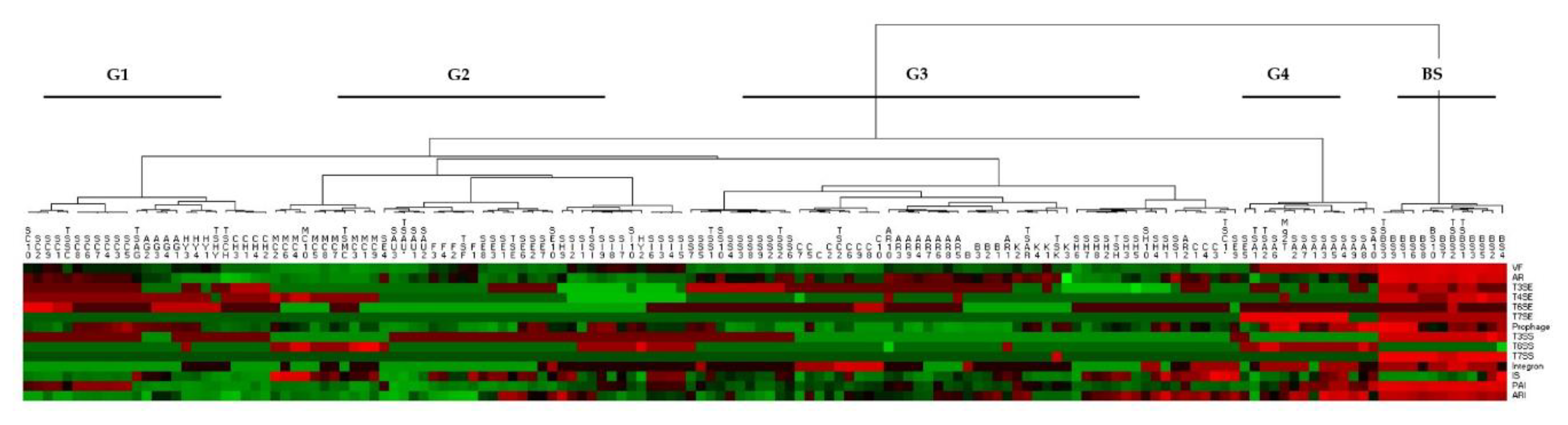
3.4. Analysis of the Distribution of Putative Virulence Elements
3.5. Virulence Factors within SAR and Species Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schleifer, K.H.; Kilpper-Bälz, R.; Devriese, L.A. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. k1oosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 1984, 5, 501–509. [Google Scholar] [CrossRef]
- Adkins, P.R.F.; Dufour, S.; Spain, J.N.; Calcutt, M.J.; Reilly, T.J.; Stewart, G.C.; Middleton, J.R. Cross-sectional study to identify staphylococcal species isolated from teat and inguinal skin of different-aged dairy heifers. J. Dairy Sci. 2018, 101, 3213–3225. [Google Scholar] [CrossRef]
- Chauhan, M.; Garlapati, V.K. Production and Characterization of a Halo-, Solvent-, Thermo-tolerant Alkaline Lipase by Staphylococcus arlettae JPBW-1, Isolated from Rock Salt Mine. Appl. Biochem. Biotechnol. 2013, 171, 1429–1443. [Google Scholar] [CrossRef]
- Pereira, E.J.; Ramaiah, N. Chromate detoxification potential of Staphylococcus sp. isolates from an estuary. Ecotoxicol. Lond. Engl. 2019, 28, 457–466. [Google Scholar] [CrossRef]
- Tu, R.-J.; Wu, H.-Y.; Lock, Y.-S.; Chen, M.-J. Evaluation of microbial dynamics during the ripening of a traditional Taiwanese naturally fermented ham. Food Microbiol. 2010, 27, 460–467. [Google Scholar] [CrossRef]
- Lavecchia, A.; Chiara, M.; Manzari, C.; Trotta, M.; Marzano, M.; Horner, D.; Pesole, G.; Placido, A. Draft Genome Sequences of Three Novel Staphylococcus arlettae Strains Isolated from a Disused Biological Safety Cabinet. Microbiol. Resour. Announc. 2018, 7. [Google Scholar] [CrossRef]
- Park, J.; Friendship, R.M.; Weese, J.S.; Poljak, Z.; Dewey, C.E. An investigation of resistance to β-lactam antimicrobials among staphylococci isolated from pigs with exudative epidermitis. BMC Vet. Res. 2013, 9, 211. [Google Scholar] [CrossRef]
- Dinakaran, V.; Shankar, M.; Jayashree, S.; Rathinavel, A.; Gunasekaran, P.; Rajendhran, J. Genome sequence of Staphylococcus arlettae strain CVD059, isolated from the blood of a cardiovascular disease patient. J. Bacteriol. 2012, 194, 6615–6616. [Google Scholar] [CrossRef]
- Bernier Gosselin, V.; Dufour, S.; Adkins, P.R.F.; Middleton, J.R. Persistence of coagulase negative staphylococcal intramammary infections in dairy goats. J. Dairy Res. 2019, 86, 211–216. [Google Scholar] [CrossRef]
- Liu, B.-H.; Lei, C.-W.; Zhang, A.-Y.; Pan, Y.; Kong, L.-H.; Xiang, R.; Wang, Y.-X.; Yang, Y.-X.; Wang, H.-N. Colocation of the Multiresistance Gene cfr and the Fosfomycin Resistance Gene fosD on a Novel Plasmid in Staphylococcus arlettae from a Chicken Farm. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Andreis, S.N.; Perreten, V.; Schwendener, S. Novel β-Lactamase blaARL in Staphylococcus arlettae. mSphere 2017, 2, e00117-17. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, D.B.; Naushad, S.; Naqvi, S.A.; Condas, L.A.Z.; Saini, V.; Kastelic, J.P.; Luby, C.; De Buck, J.; Barkema, H.W. Prevalence and Genetic Basis of Antimicrobial Resistance in Non-aureus Staphylococci Isolated from Canadian Dairy Herds. Front. Microbiol. 2018, 9, 256. [Google Scholar] [CrossRef]
- Xu, J.; Tan, X.; Zhang, X.; Xia, X.; Sun, H. The diversities of staphylococcal species, virulence and antibiotic resistance genes in the subclinical mastitis milk from a single Chinese cow herd. Microb. Pathog. 2015, 88, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Qu, T.-T.; Yang, Q.; Shen, P.; Wei, Z.-Q.; Yu, Y.-S. Novel Vancomycin-Resistance Transposon, Plasmid Replicon Types, and Virulence Factors of Vancomycin-Resistant Enterococci in Zhejiang, China. Microb. Drug Resist. 2012, 18, 183–188. [Google Scholar] [CrossRef]
- Xu, X.; Chen, C.; Lin, D.; Guo, Q.; Hu, F.; Zhu, D.; Li, G.; Wang, M. The Fosfomycin Resistance Gene fosB3 Is Located on a Transferable, Extrachromosomal Circular Intermediate in Clinical Enterococcus faecium Isolates. PLoS ONE 2013, 8, e78106. [Google Scholar] [CrossRef]
- Schnabel, E.L.; Jones, A.L. Distribution of Tetracycline Resistance Genes and Transposons among Phylloplane Bacteria in Michigan Apple Orchards. Appl. Environ. Microbiol. 1999, 65, 4898–4907. [Google Scholar]
- Li, M.; Guan, M.; Jiang, X.F.; Yuan, F.Y.; Xu, M.; Zhang, W.Z.; Lu, Y. Genetic polymorphism of the accessory gene regulator (agr) locus in Staphylococcus epidermidis and its association with pathogenicity. J. Med. Microbiol. 2004, 53, 545–549. [Google Scholar] [CrossRef]
- Vandenesch, F.; Projan, S.J.; Kreiswirth, B.; Etienne, J.; Novick, R.P. Agr-related sequences in Staphylococcus lugdunensis. FEMS Microbiol. Lett. 1993, 111, 115–122. [Google Scholar] [CrossRef]
- Choo, E.J.; Chambers, H.F. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 2016, 48, 267–273. [Google Scholar] [CrossRef]
- Chiara, M.; Placido, A.; Picardi, E.; Ceci, L.R.; Horner, D.S.; Pesole, G. A-GAME: Improving the assembly of pooled functional metagenomics sequence data. BMC Genom. 2018, 19, 44. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Lamers, R.P.; Muthukrishnan, G.; Castoe, T.A.; Tafur, S.; Cole, A.M.; Parkinson, C.L. Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol. Biol. 2012, 12, 171. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, J.; Tai, C.; Deng, Z.; Zhong, W.; He, Y.; Ou, H.-Y. VRprofile: Gene-cluster-detection-based profiling of virulence and antibiotic resistance traits encoded within genome sequences of pathogenic bacteria. Brief. Bioinform. 2018, 19, 566–574. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Caraux, G.; Pinloche, S. PermutMatrix: A graphical environment to arrange gene expression profiles in optimal linear order. Bioinform. Oxf. Engl. 2005, 21, 1280–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mašlaňová, I.; Wertheimer, Z.; Sedláček, I.; Švec, P.; Indráková, A.; Kovařovic, V.; Schumann, P.; Spröer, C.; Králová, S.; Šedo, O.; et al. Description and Comparative Genomics of Macrococcus caseolyticus subsp. hominis subsp. nov., Macrococcus goetzii sp. nov., Macrococcus epidermidis sp. nov., and Macrococcus bohemicus sp. nov., Novel Macrococci From Human Clinical Material With Virulence Potential and Suspected Uptake of Foreign DNA by Natural Transformation. Front. Microbiol. 2018, 9, 1178. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, S.; Hill, C.; McAuliffe, O. The Genus Macrococcus: An Insight into Its Biology, Evolution, and Relationship With Staphylococcus. Adv. Appl. Microbiol. 2018, 105, 1–50. [Google Scholar] [CrossRef]
- Alcalde-Rico, M.; Hernando-Amado, S.; Blanco, P.; Martínez, J.L. Multidrug Efflux Pumps at the Crossroad between Antibiotic Resistance and Bacterial Virulence. Front. Microbiol. 2016, 7, 1483. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K. Multidrug resistance pumps in bacteria: Variations on a theme. Trends Biochem. Sci. 1994, 19, 119–123. [Google Scholar] [CrossRef]
- Vargiu, A.V.; Pos, K.M.; Poole, K.; Nikaido, H. Editorial: Bad Bugs in the XXIst Century: Resistance Mediated by Multi-Drug Efflux Pumps in Gram-Negative Bacteria. Front. Microbiol. 2016, 7, 833. [Google Scholar] [CrossRef] [Green Version]
- Brambila-Tapia, A.J.L.; Armenta-Medina, D.; Rivera-Gomez, N.; Perez-Rueda, E. Main Functions and Taxonomic Distribution of Virulence Genes in Brucella melitensis 16 M. PLoS ONE 2014, 9, e100349. [Google Scholar] [CrossRef] [Green Version]
- Jeukens, J.; Freschi, L.; Vincent, A.T.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Charette, S.J.; Levesque, R.C. A Pan-Genomic Approach to Understand the Basis of Host Adaptation in Achromobacter. Genome Biol. Evol. 2017, 9, 1030–1046. [Google Scholar] [CrossRef]
- Argemi, X.; Hansmann, Y.; Prola, K.; Prévost, G. Coagulase-Negative Staphylococci Pathogenomics. Int. J. Mol. Sci. 2019, 20, 1215. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Antibiotic Tested | In Vitro Susceptibility | Predicted Genes | |
|---|---|---|---|---|
| 1. | β-Lactam | Oxacillin | R | ampS, ampC, gob-2, gob-18, imp-35, arl-1, nmcA, nmcR, mecD, mecI, mecR1 |
| 2. | Ampicillin | R | ||
| 3. | Cefoxitin | R | ||
| 4. | Ceftaroline | R | ||
| 5. | Imipenem | R | ||
| 6. | Penicillin | R | ||
| 7. | Lincosamide | Clindamycin | R | ermC, ermK, cfrC, oleC, mexL, mexS |
| 8. | Macrolide | Erythromycin | R | |
| 9. | - | Fosfomycin | R | fosD, fosB5, fosA6, fosB2 |
| 10. | Fusidic Acid | Fusidic Acid | R | fusA |
| 11. | Fluoroquinolone | Ciprofloxacin | S | gyrA, gyrB, norA, norB, pmrA, patA, arlS, arlR |
| 12. | Moxifloxacin | S | ||
| 13. | Peptide | Daptomycin | S | cls, pgsA, mprf |
| 14. | Glycopeptide | Teicoplanin | S | - |
| 15. | Vancomycin | S | vanKI, vanK, vanTG, vanHM, vanH, vanT, vanL | |
| 16. | Tetracycline | Tetracycline | S | emrA, emrB, emrY, emrR, tet(X3), tetT, tet35, tet47, adeN, adeL, adeR, |
| 17. | Tigecycline | S | ||
| 18. | Aminoglycoside | Gentamicin | S | aac(6′), aph(2″), mexS, mexL, acrS |
| 19. | Oxazolidinone | Linezolid | S | cfrB, cfrC, lmrS, poxtA |
| 20. | Diaminopyrimidine-Sulfonamide | Trimethoprim-Sulfamethoxazole | S | dfr |
| 21. | Monoxycarbolic Acid | Mupirocin | S | mup, ileS |
| 22. | Nitrofuran | Nitrofurantoin | S | - |
| 23. | Rifamycin | Rifampin | S | rpoB2 |
| Strain * | N | Antibiotic Efflux | Antibiotic Inactivation | Antibiotic Target Alteration | Antibiotic Target Protection | Antibiotic Target Replacement |
|---|---|---|---|---|---|---|
| B | 227 | 117 | 39 | 57 | 10 | 4 |
| TS | 227 | 119 | 37 | 55 | 11 | 5 |
| B1 | 224 | 117 | 35 | 53 | 14 | 5 |
| B2 | 219 | 116 | 33 | 51 | 14 | 5 |
| B3 | 215 | 116 | 29 | 52 | 13 | 5 |
| AR1 | 219 | 113 | 32 | 59 | 9 | 6 |
| AR2 | 215 | 114 | 30 | 54 | 12 | 5 |
| AR3 | 220 | 114 | 33 | 56 | 12 | 5 |
| AR4 | 225 | 120 | 34 | 54 | 12 | 5 |
| AR5 | 216 | 113 | 32 | 54 | 12 | 5 |
| AR6 | 215 | 112 | 30 | 57 | 11 | 5 |
| AR7 | 220 | 113 | 32 | 58 | 12 | 5 |
| AR8 | 220 | 114 | 33 | 56 | 12 | 5 |
| AR9 | 223 | 116 | 35 | 56 | 11 | 5 |
| AR10 | 228 | 118 | 34 | 58 | 13 | 5 |
| AR11 | 225 | 119 | 32 | 57 | 12 | 5 |
| AR12 | 212 | 113 | 30 | 53 | 11 | 5 |
| AR13 | 219 | 112 | 31 | 59 | 12 | 5 |
| AR14 | 215 | 113 | 33 | 53 | 11 | 5 |
| AR15 | 234 | 125 | 35 | 57 | 12 | 5 |
| AR16 | 222 | 115 | 35 | 55 | 12 | 5 |
| AR17 | 217 | 114 | 35 | 55 | 8 | 5 |
| AR18 | 218 | 114 | 33 | 55 | 10 | 6 |
| Vfclass | Virulence Factors | Related Genes | Hits |
|---|---|---|---|
| 1-Enzymes | Lipase | lip | PROKKA_00412 |
| Serine V8 protease | sspA | PROKKA_02492 | |
| Thermonuclease | nuc | PROKKA_01673 | |
| 2-Immune evasion | Capsule | undetermined | PROKKA_00763; PROKKA_01266; PROKKA_01584; PROKKA_02340; PROKKA_02341; PROKKA_02342; PROKKA_02343; PROKKA_02439; PROKKA_02440; PROKKA_02511; |
| Capsule (Acinetobacter) | - | PROKKA_02505 | |
| Polyglutamic acid capsule (Bacillus) | capB | PROKKA_01886 | |
| capC | PROKKA_01887 | ||
| Polysaccharide capsule (Bacillus) | galE | PROKKA_01613 | |
| 3-Antiphagocytosis | Capsule (Klebsiella) | uge | PROKKA_02504 |
| 4-Nutritional factor | Allantoin utilization (Klebsiella) | - | PROKKA_00370 |
| PROKKA_00367 | |||
| PROKKA_00364; PROKKA_00365 | |||
| 5-Serum resistance and immune evasion | LPS (Francisella) | wbtP | PROKKA_02442 |
| Strains *. | N | Genes |
|---|---|---|
| B1; B2; B3; AR1; AR10; AR11; AR12; AR13; AR14; AR15; AR16; AR17; AR18; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9; TS; B | 2 | Undetermined capsule, sspA |
| B1; B2; B3; AR1; AR10; AR11; AR12; AR14; AR15; AR16; AR17; AR18; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9; TS; B | 1 | lip |
| B1; B2; B3; AR10; AR11; AR12; AR13; AR14; AR15; AR16; AR17; AR18; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9; TS; B | 1 | nuc |
| B1; B2; B3; AR1; AR10; AR11; AR12; AR13; AR14; AR15; AR16; AR17; AR18; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9; B | 3 | capC, capB, galE |
| AR1; AR10; AR11; AR13; AR14; AR15; AR16; AR17; AR18; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9; B | 1 | wbtP |
| AR10; AR11; AR12; AR13; AR14; AR15; AR16; AR2; AR3; AR4; AR5; AR6; AR7; AR8; AR9 | 6 | esaB, esaA, esBA, ess, essC, essA |
| B2; AR10; AR13; AR3; AR7; AR8; AR9 | 1 | lspA |
| B2; AR12; AR13; AR18; AR7; AR8 | 1 | gtaB |
| AR12; AR13; AR7; AR8 | 2 | tuf, katA |
| B1; B2; B3 | 1 | vctC |
| B2; AR12; AR8 | 5 | plr/gapA, ndk, eno, acpBL, flmH |
| B2; AR13; AR8 | 2 | lgt, lpeA |
| AR1; AR15; AR8 | 1 | cylR2 |
| AR12; AR13; AR8 | 1 | slrA |
| AR17; B | 1 | uge |
| B1; AR17 | 2 | icaB, icaA |
| B2; AR8 | 1 | lisR |
| B2; AR12 | 1 | gnd |
| B2; AR13 | 1 | sigA/rpoV |
| B3; B | 1 | Allantoin utilization |
| AR8 | 2 | groEL, lplA1 |
| AR12 | 1 | hemL |
| AR17 | 1 | icaC |
| Species * | N | Genes |
|---|---|---|
| BS; MC; SA; SAG; SAR; SAU; SC; SCH; SE; SF; SH; SHY; SK; SS; SSC; SSI | 1 | Capsule Undetermined |
| SAG; SAR; SAU; SC; SCH; SE; SF; SH; SHY; SK; SS; SSI | 1 | nuc |
| BS; SAG; SAR; SAU; SC; SCH; SE; SH; SHY; SK; SS; SSI | 2 | capB; capC |
| BS; MC; SAG; SAR; SC; SCH; SE; SF; SHY; SK; SS | 1 | vctC |
| SA; SAR; SC; SCH; SE; SF; SH; SK; SS | 1 | lip |
| MC; SA; SAR; SC; SE; SK; SS; SSC; SSI | 1 | icaA |
| BS; MC; SAG; SAR; SCH; SF; SHY; SS; SSC | 1 | lgt |
| BS; SAR; SAU; SC; SF; SHY; SK; SS; SSI | 1 | galE |
| SA; SAR; SC; SE; SK; SS; SSC; SSI | 1 | icaB |
| MC; SAG; SAR; SC; SH; SHY; SK; SSI | 1 | wbtP |
| BS; SA; SAG; SAR; SE; SHY; SSI | 1 | essC |
| SA; SAR; SC; SE; SK; SS; SSC | 1 | sspA |
| MC; SAR; SAU; SC; SF; SH; SSI | 1 | cylR2 |
| SA; SAG; SAR; SE; SHY; SSI | 3 | esaB; essB; esxA |
| MC; SAG; SAR; SCH; SHY; SSC | 1 | lisR |
| BS; SAG; SAR; SHY; SK; SSI | 1 | lspA |
| MC; SAR; SC; SH; SHY; SSI | 1 | uge |
| BS; SAR; SH; SHY; SK; SSI | 1 | Capsule (Acinetobacter) |
| SA; SAG; SAR; SE; SSI | 1 | esaA |
| SAR; SC; SHY; SSC; SSI | 1 | Allantoin utilization (Klebsiella) |
| BS; MC; SAR; SF; SS | 1 | gtaB |
| MC; SAR; SC; SSI | 1 | LPS O-antigen (P. aeruginosa) |
| BS; MC; SAR; SSC | 1 | ndk |
| SA; SAR; SE | 1 | essA |
| BS; SAR; SAU | 1 | Capsule (Enterococcus) |
| BS; SAR; SK | 1 | LPS rfb locus (Klebsiella) |
| BS; MC; SAR | 1 | lplA1 |
| SAR; SAU | 1 | lpeA |
| SAR; SE | 1 | eno |
| BS; SAR | 4 | plr/gapA; katA; acpXL; gnd |
| SAR | 6 | flmH; T6SS-II(Klebsiella); tuf; groEL; slrA; sigA/rpoV |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavecchia, A.; Chiara, M.; De Virgilio, C.; Manzari, C.; Monno, R.; De Carlo, A.; Pazzani, C.; Horner, D.; Pesole, G.; Placido, A. Staphylococcus arlettae Genomics: Novel Insights on Candidate Antibiotic Resistance and Virulence Genes in an Emerging Opportunistic Pathogen. Microorganisms 2019, 7, 580. https://doi.org/10.3390/microorganisms7110580
Lavecchia A, Chiara M, De Virgilio C, Manzari C, Monno R, De Carlo A, Pazzani C, Horner D, Pesole G, Placido A. Staphylococcus arlettae Genomics: Novel Insights on Candidate Antibiotic Resistance and Virulence Genes in an Emerging Opportunistic Pathogen. Microorganisms. 2019; 7(11):580. https://doi.org/10.3390/microorganisms7110580
Chicago/Turabian StyleLavecchia, Anna, Matteo Chiara, Caterina De Virgilio, Caterina Manzari, Rosa Monno, Armando De Carlo, Carlo Pazzani, David Horner, Graziano Pesole, and Antonio Placido. 2019. "Staphylococcus arlettae Genomics: Novel Insights on Candidate Antibiotic Resistance and Virulence Genes in an Emerging Opportunistic Pathogen" Microorganisms 7, no. 11: 580. https://doi.org/10.3390/microorganisms7110580
APA StyleLavecchia, A., Chiara, M., De Virgilio, C., Manzari, C., Monno, R., De Carlo, A., Pazzani, C., Horner, D., Pesole, G., & Placido, A. (2019). Staphylococcus arlettae Genomics: Novel Insights on Candidate Antibiotic Resistance and Virulence Genes in an Emerging Opportunistic Pathogen. Microorganisms, 7(11), 580. https://doi.org/10.3390/microorganisms7110580