Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis

 , ,
, , 
Abstract
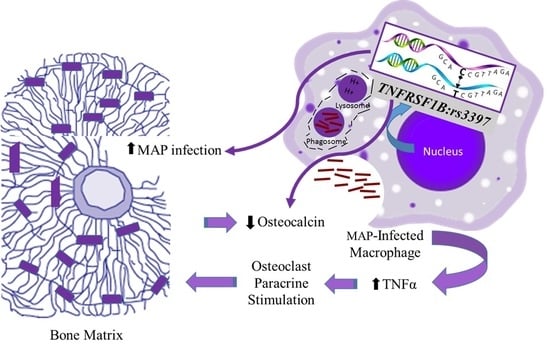
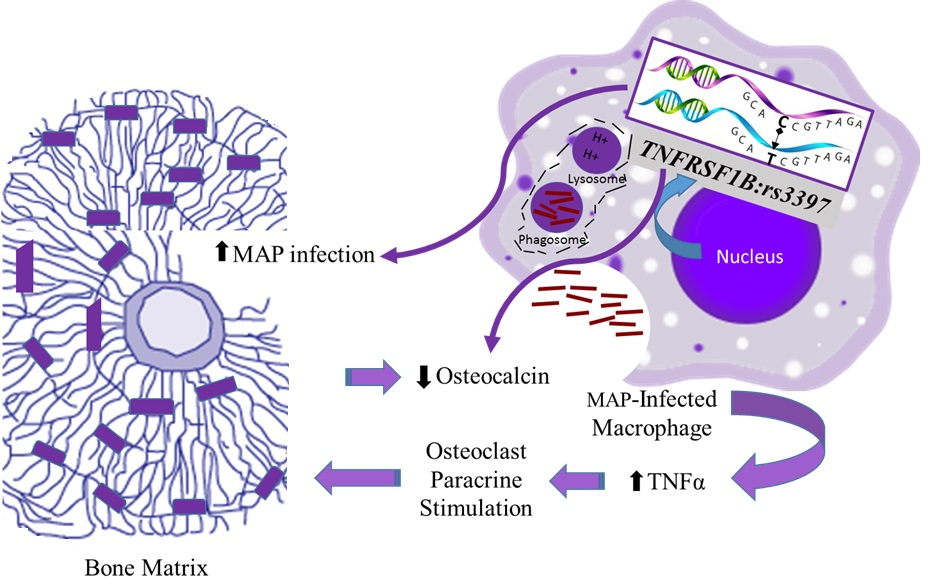
:
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Detection of MAP Infection in the Blood of RA Patients by nPCR
2.3. Measurement of Active Osteocalcin Level
2.4. Identification of SNPs in TNFα, TNFRSF1A, and TNFRSF1B
2.5. RNA Extraction and Measurement of TNFα, TNFRSF1A, and TNFRSF1B Expression
2.6. Statistical Analysis
3. Results
3.1. MAP DNA Detected in RA Patients
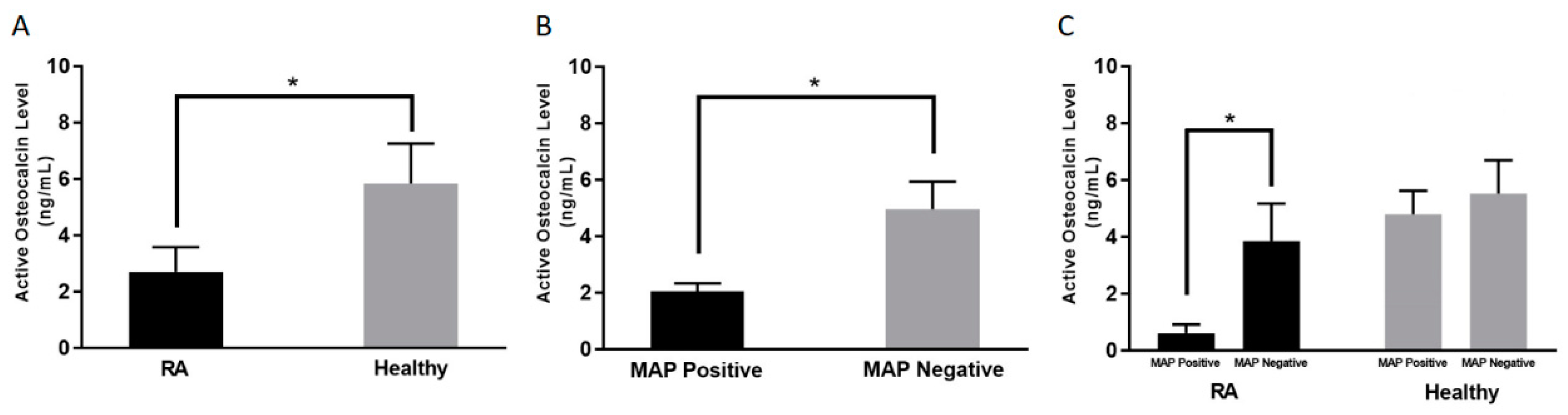
3.2. Lower Active Osteocalcin Levels in RA Patients
3.3. Lower Active Osteocalcin Level is Detected in MAP Positive RA Patients
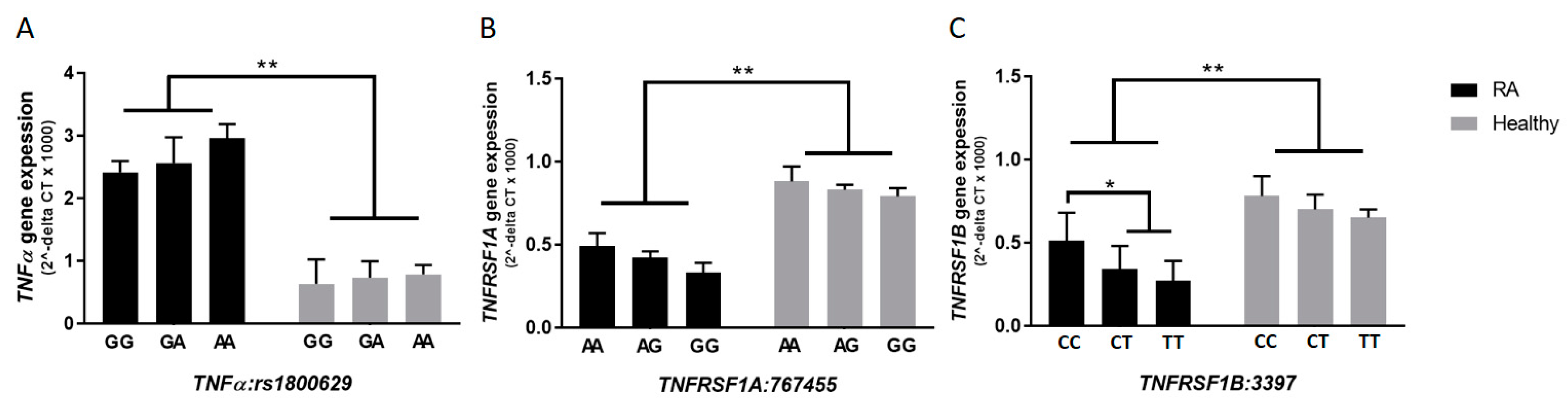
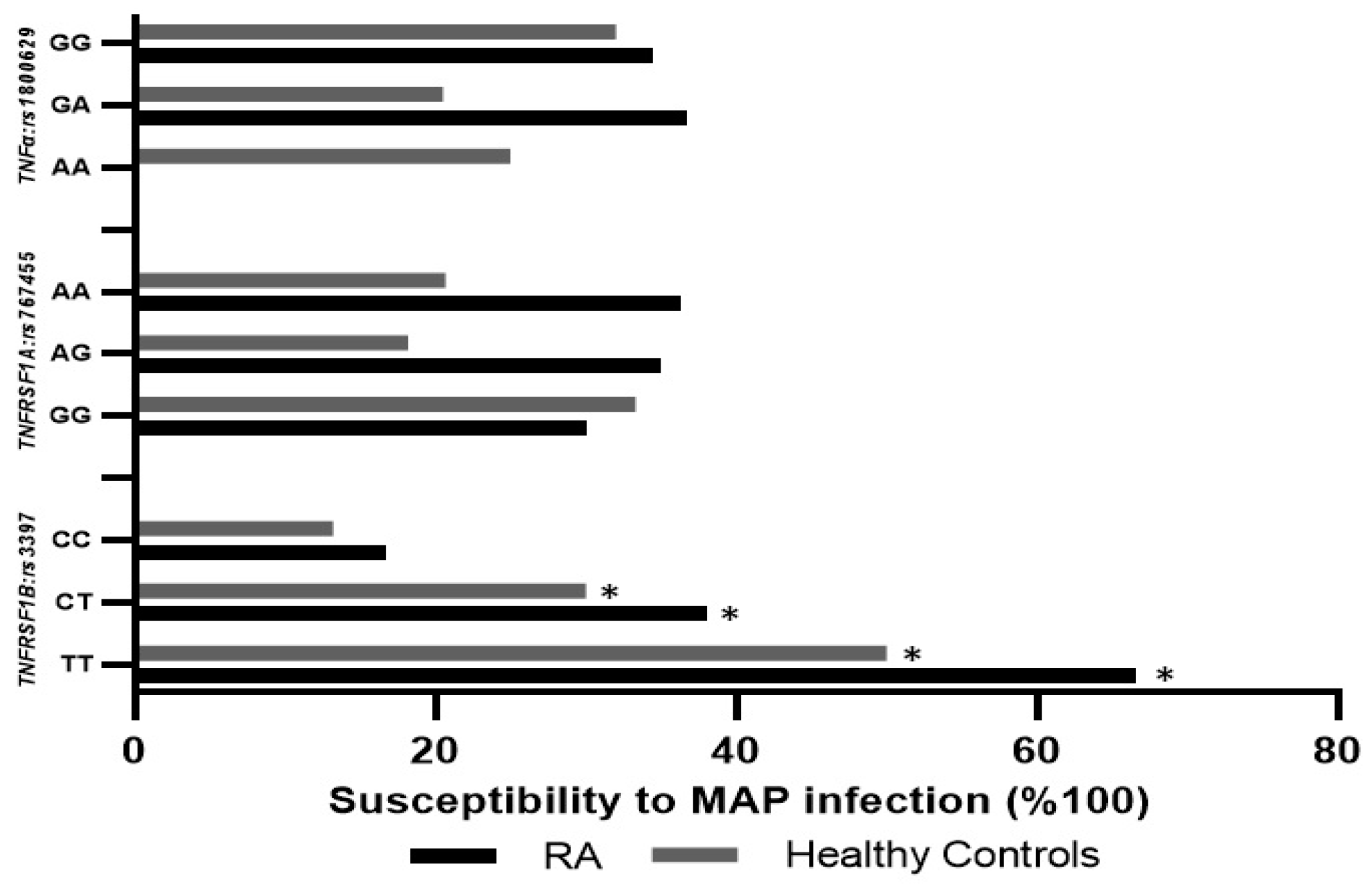
3.4. Frequency of SNPs in TNFα, TNFRSF1A, and TNFRSF1B in RA Patients
3.5. rs3397 Downregulates TNFRSF1B Expression in RA Patients
3.6. TNFRSF1B:rs3397 is Associated with Lower Active Osteocalcin Level in RA Patients
3.7. TNFRSF1B:rs3397 is Associated with MAP Infection in RA
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, N.E.; Pressman, A.R.; Star, V.L.; Cummings, S.R.; Nevitt, M.C. Rheumatoid arthritis and bone mineral density in elderly women. J. Bone Miner. Res. 1995, 10, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hooyman, J.R.; Joseph Melton, L., III; Nelson, A.M.; O’Fallon, W.M.; Riggs, B.L. Fractures after rheumatoid arthritis a population-based study. Arthritis Rheum. 1984, 27, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Westhovens, R.; Nijs, J.; Taelman, V.; Dequeker, J. Body composition in rheumatoid arthritis. Br. J. Rheumatol. 1997, 36, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodder, M.C.; de Jong, Z.; Kostense, P.J.; Molenaar, E.T.; Staal, K.; Voskuyl, A.E.; Lems, W.F. Bone mineral density in patients with rheumatoid arthritis: Relation between disease severity and low bone mineral density. Ann. Rheum. Dis. 2004, 63, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Spector, T.D.; Hall, G.M.; McCloskey, E.V.; Kanis, J.A. Risk of vertebral fracture in women with rheumatoid arthritis. BMJ: Br. Med. J. 1993, 306, 558. [Google Scholar] [CrossRef] [Green Version]
- Saville, P.D.; Kharmosh, O. Osteoporosis of rheumatoid arthritis: Influence of age, sex and corticosteroids. Arthritis Rheum. 1967, 10, 423–430. [Google Scholar] [CrossRef]
- Cooper, C.; Coupland, C.; Mitchell, M. Rheumatoid arthritis, corticosteroid therapy and hip fracture. Ann. Rheum. Dis. 1995, 54, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Manolagas, S.C.; Jilka, R.L. Bone marrow, cytokines, and bone remodeling—Emerging insights into the pathophysiology of osteoporosis. New Engl. J. Med. 1995, 332, 305–311. [Google Scholar] [CrossRef]
- Gough, A.; Sambrook, P.; Devlin, J.; Huissoon, A.; Njeh, C.; Robbins, S.; Emery, P. Osteoclastic activation is the principal mechanism leading to secondary osteoporosis in rheumatoid arthritis. J. Rheumatol. 1998, 25, 1282–1289. [Google Scholar]
- Naser, A.; Qasem, A.; Naser, S.A. Mycobacterial infection influences bone biomarker levels in patients with Crohn’s disease. Can. J. Physiol. Pharmacol. 2018, 96, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6, e000246. [Google Scholar] [CrossRef] [PubMed]
- Sharp, R.C.; Beg, S.A.; Naser, S.A. Polymorphisms in protein tyrosine phosphatase non-receptor type 2 and 22 (PTPN2/22) are linked to hyper-proliferative T-Cells and susceptibility to mycobacteria in rheumatoid arthritis. Front. Cell. Infect. Microbiol. 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Qasem, A.; Abdel-Aty, A.; Abu-Suwa, H.; Naser, S.A. Oxidative stress due to Mycobacterium avium subspecies paratuberculosis (MAP) infection upregulates selenium-dependent GPx activity. Gut Pathog. 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, R.C.; Naser, E.S.; Alcedo, K.P.; Qasem, A.; Abdelli, L.S.; Naser, S.A. Development of multiplex PCR and multi-color fluorescent in situ hybridization (m-FISH) coupled protocol for detection and imaging of multi-pathogens involved in inflammatory bowel disease. Gut Pathog. 2018, 10, 51. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: Ncbi.nlm.nih.gov (accessed on 5 April 2019).
- Xu, S.; Wang, Y.; Lu, J.; Xu, J. Osteoprotegerin and RANKL in the pathogenesis of rheumatoid arthritis-induced osteoporosis. Rheumatol. Int. 2012, 32, 3397–3403. [Google Scholar] [CrossRef]
- Johnell, O.; Kanis, J.A. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos. Int. 2006, 17, 1726–1733. [Google Scholar] [CrossRef]
- Peris, P. Stress fractures in rheumatological practice: Clinical significance and localizations. Rheumatol. Int. 2002, 22, 77–79. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Manning, C.; Tsay, A.; Naito, A.; Pan, C.; Amento, E.; Goldring, S.R. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000, 43, 250–258. [Google Scholar] [CrossRef]
- Bekker, P.J.; Holloway, D.; Nakanishi, A.; Arrighi, H.M.; Dunstan, C.R. Osteoprotegerin (OPG) has potent and sustained anti-resorptive activity in postmenopausal women. J. Bone Miner. Res. 1999, 14, S180. [Google Scholar]
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef]
- Shigeyama, Y.; Pap, T.; Kunzler, P.; Simmen, B.R.; Gay, R.E.; Gay, S. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 2523–2530. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Goldring, S.R. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 2143–2151. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Heufelder, A.E. The role of osteoprotegerin and receptor activator of nuclear factor κB ligand in the pathogenesis and treatment of rheumatoid arthritis. Arthritis Rheum. 2001, 44, 253–259. [Google Scholar] [CrossRef]
- Qasem, A.; Naser, S.A. TNFα inhibitors exacerbate Mycobacterium paratuberculosis infection in tissue culture: A rationale for poor response of patients with Crohn’s disease to current approved therapy. BMJ Open Gastroenterol. 2018, 5, e000216. [Google Scholar] [CrossRef] [Green Version]
- Blumenthal, A.; Lauber, J.; Hoffmann, R.; Ernst, M.; Keller, C.; Buer, J.; Reiling, N. Common and unique gene expression signatures of human macrophages in response to four strains of Mycobacterium avium that differ in their growth and persistence characteristics. Infect. Immun. 2005, 73, 3330–3341. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.J.; Evanson, O.A.; Deng, M.; Abrahamsen, M.S. Gene expression and antimicrobial activity of bovine macrophages in response to Mycobacterium avium subsp. paratuberculosis. Vet. Pathol. 2004, 41, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.J.; Evanson, O.A.; Deng, M.; Abrahamsen, M.S. Sequential patterns of gene expression by bovine monocyte-derived macrophages associated with ingestion of mycobacterial organisms. Microb. Pathog. 2004, 37, 215–224. [Google Scholar] [CrossRef]
- Murphy, J.T.; Sommer, S.; Kabara, E.A.; Verman, N.; Kuelbs, M.A.; Saama, P.; Coussens, P.M. Gene expression profiling of monocyte-derived macrophages following infection with Mycobacterium avium subspecies avium and Mycobacterium avium subspecies paratuberculosis. Physiol. Genom. 2006, 28, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.L.; Qasem, A.; Sharp, R.C.; Abdelli, L.S.; Naser, S.A. Systematic review and meta-analysis on the association of tuberculosis in Crohn’s disease patients treated with tumor necrosis factor-α inhibitors (Anti-TNFα). World J. Gastroenterol. 2018, 24, 2764. [Google Scholar] [CrossRef]
- Qasem, A.; Naser, A.E.; Naser, S.A. The alternate effects of anti-TNFα therapeutics and their role in mycobacterial granulomatous infection in Crohn’s disease. Expert Rev. Anti-Infect. Ther. 2017, 15, 637–643. [Google Scholar] [CrossRef]
- Qasem, A.; Safavikhasraghi, M.; Naser, S.A. A single capsule formulation of RHB-104 demonstrates higher anti-microbial growth potency for effective treatment of Crohn’s disease associated with Mycobacterium avium subspecies paratuberculosis. Gut Pathog. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, J.C.J.; Flynn, J.L.; Kirschner, D.E. Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J. Immunol. 2009, 182, 3706–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigeler, A.; Chromik, A.M.; Geisler, A.; Bulut, D.; Hilgert, C.; Krieg, A.; Mittelkötter, U. Synergistic apoptotic effects of taurolidine and TRAIL on squamous carcinoma cells of the esophagus. Int. J. Oncol. 2008, 32, 1205–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Lee, H.S.; Yoon, J.H.; Kim, C.Y.; Park, M.H.; Kim, L.; Shin, H.D. Association of TNF-α promoter polymorphisms with the clearance of hepatitis B virus infection. Hum. Mol. Genet. 2003, 12, 2541–2546. [Google Scholar] [CrossRef] [Green Version]
- Ok, M.; Einsele, H.; Loeffler, J. Genetic susceptibility to Aspergillus fumigatus infections. Int. J. Med Microbiol. 2011, 301, 445–452. [Google Scholar] [CrossRef]
- Alkoky, H.; Qadir, G.; Etomi, O.; Tahir, H.; Pakozdi, A. AB0352 Significance of occult infections in inflammatory arthritis patients receiving biologic therapies in east London. Ann. Rheum. Dis. 2018. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Number | Age Range | Average Age | Gender Ratio (Male/Female %) |
|---|---|---|---|---|
| All Subjects | 102 | 21–75 | 39 | 33:67 |
| Rheumatoid Arthritis | 54 | 22–75 | 49 | 25:75 |
| Healthy Controls | 48 | 20–63 | 30 | 41:59 |
| Gene | Reference SNP | Gene Mutation * | Location and AA Change * | Mutation Phenotype |
|---|---|---|---|---|
| TNF | rs1800629 | G > A | Promoter | Higher susceptibility to develop CD |
| TNFRSF1A | rs767455 | C > T | Exon 4 (R > Q) | Poor response to anti-TNFα treatment in CD |
| TNFRSF1B | rs3397 | C > T | Exon 10 (N/A) | Higher susceptibility to MAP infection in CD |
| Genotype | RA Patients (n = 48) | Healthy Controls (n = 54) | p-value * | OR | 95% CI |
|---|---|---|---|---|---|
| TNFα:rs1800629 | |||||
| GG (Reference allele) | 29 (60%) | 44 (82%) | 0.03 | ||
| GA | 19 (40%) | 8 (15%) | 0.02 | 3.6 | 1.37–9.54 |
| AA | 0 (0%) | 2 (0%) | 0.23 | 0.2 | 0.01–3.36 |
| GA + AA | 19 (40%) | 10 (19%) | 0.04 | 2.9 | 1.17–7.07 |
| TNFRSF1A:rs767455 | |||||
| AA (Reference allele) | 22 (46%) | 29 (54%) | 0.42 | ||
| AG | 20 (42%) | 22 (41%) | 0.47 | 1.2 | 0.53–2.73 |
| GG | 6 (12%) | 3 (6%) | 0.17 | 2.6 | 0.59–11.7 |
| AG + GG | 26 (54%) | 25 (46%) | 0.31 | 1.4 | 0.63–2.99 |
| TNFRSF1B:rs3397 | |||||
| CC (Reference allele) | 18 (37%) | 38 (70%) | 0.02 | ||
| CT | 21 (44%) | 10 (19%) | 0.03 | 4.43 | 1.73–11.3 |
| TT | 9 (19%) | 6 (11%) | 0.29 | 3.17 | 0.98–10.3 |
| CT + TT | 30 (62%) | 16 (30%) | 0.03 | 4 | 1.73–9.05 |
| Treatment Group | Number of Patients Receiving Treatment | MAP Infection | Average Osteocalcin (ng/mL) |
|---|---|---|---|
| Anti-TNFα | 20 | 6/20 (30.0%) | 2.71 ± 0.82 |
| Methotrexate | 28 | 10/28 (35.7%) | 2.49 ± 0.57 |
| Corticosteroids | 13 | 3/13 (23.1%) | 2.98 ± 0.69 |
| Hydroxychloroquine | 9 | 4/9 (44.4%) | 2.23 ± 0.26 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naser, A.; Odeh, A.K.; Sharp, R.C.; Qasem, A.; Beg, S.; Naser, S.A. Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis. Microorganisms 2019, 7, 646. https://doi.org/10.3390/microorganisms7120646
Naser A, Odeh AK, Sharp RC, Qasem A, Beg S, Naser SA. Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis. Microorganisms. 2019; 7(12):646. https://doi.org/10.3390/microorganisms7120646
Chicago/Turabian StyleNaser, Amna, Ahmad K. Odeh, Robert C. Sharp, Ahmad Qasem, Shazia Beg, and Saleh A. Naser. 2019. "Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis" Microorganisms 7, no. 12: 646. https://doi.org/10.3390/microorganisms7120646
APA StyleNaser, A., Odeh, A. K., Sharp, R. C., Qasem, A., Beg, S., & Naser, S. A. (2019). Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis. Microorganisms, 7(12), 646. https://doi.org/10.3390/microorganisms7120646