Root-Associated Endophytic Bacterial Community Composition of Pennisetum sinese from Four Representative Provinces in China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Genomic DNA Extraction and PCR Amplification
2.3. Library Construction and Sequencing
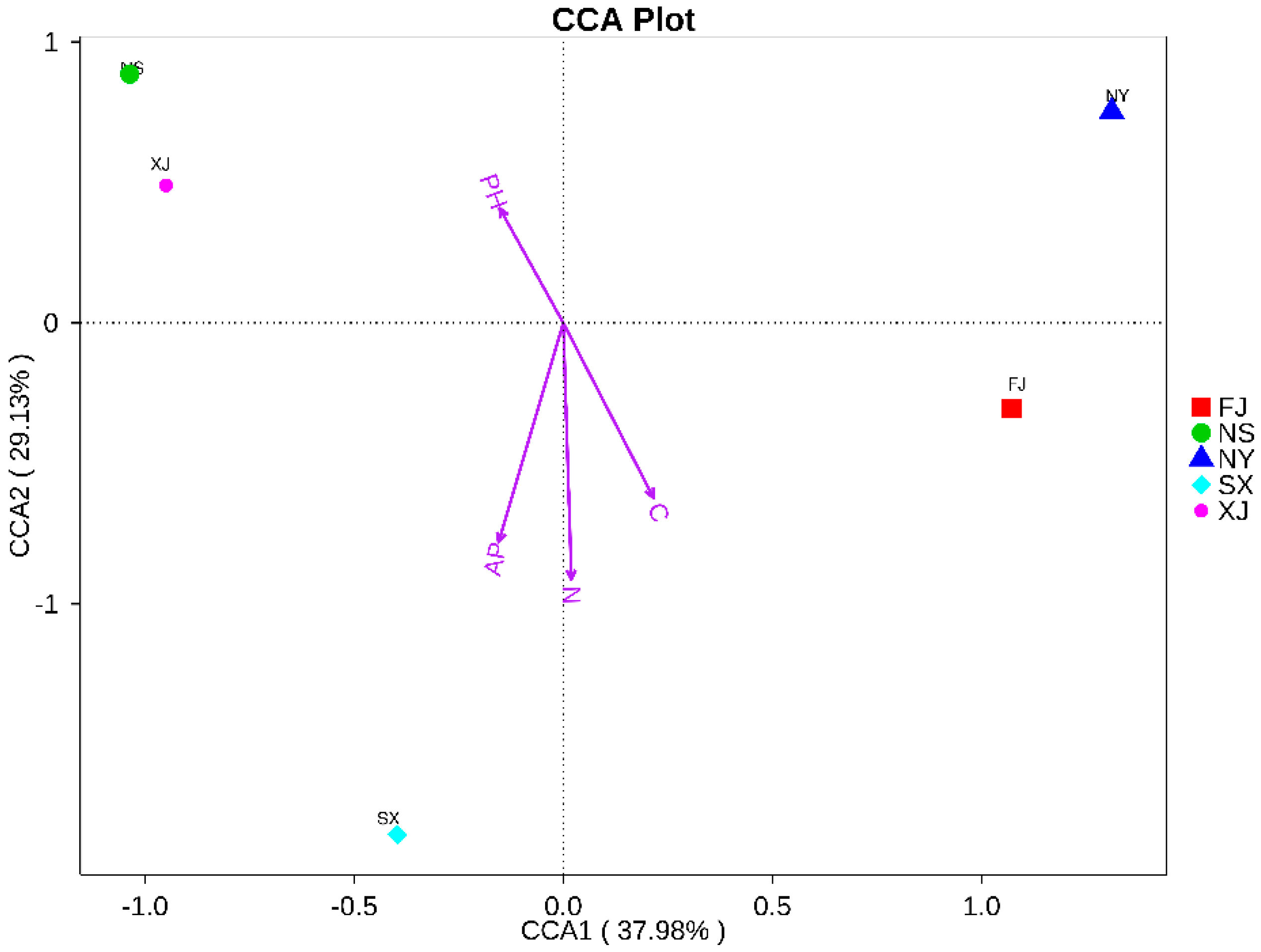
2.4. Statistical Analysis
3. Results
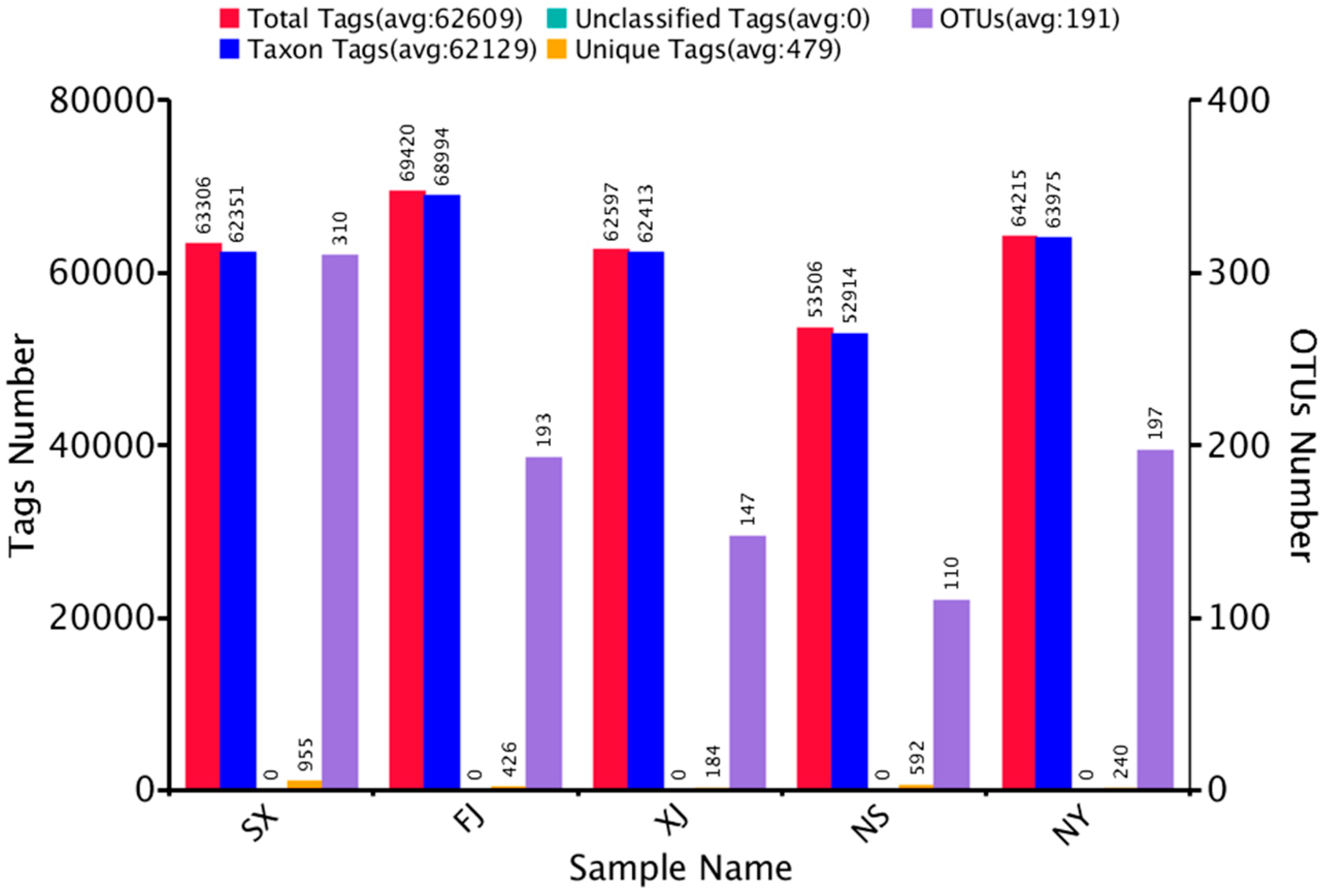
3.1. Sequencing Results
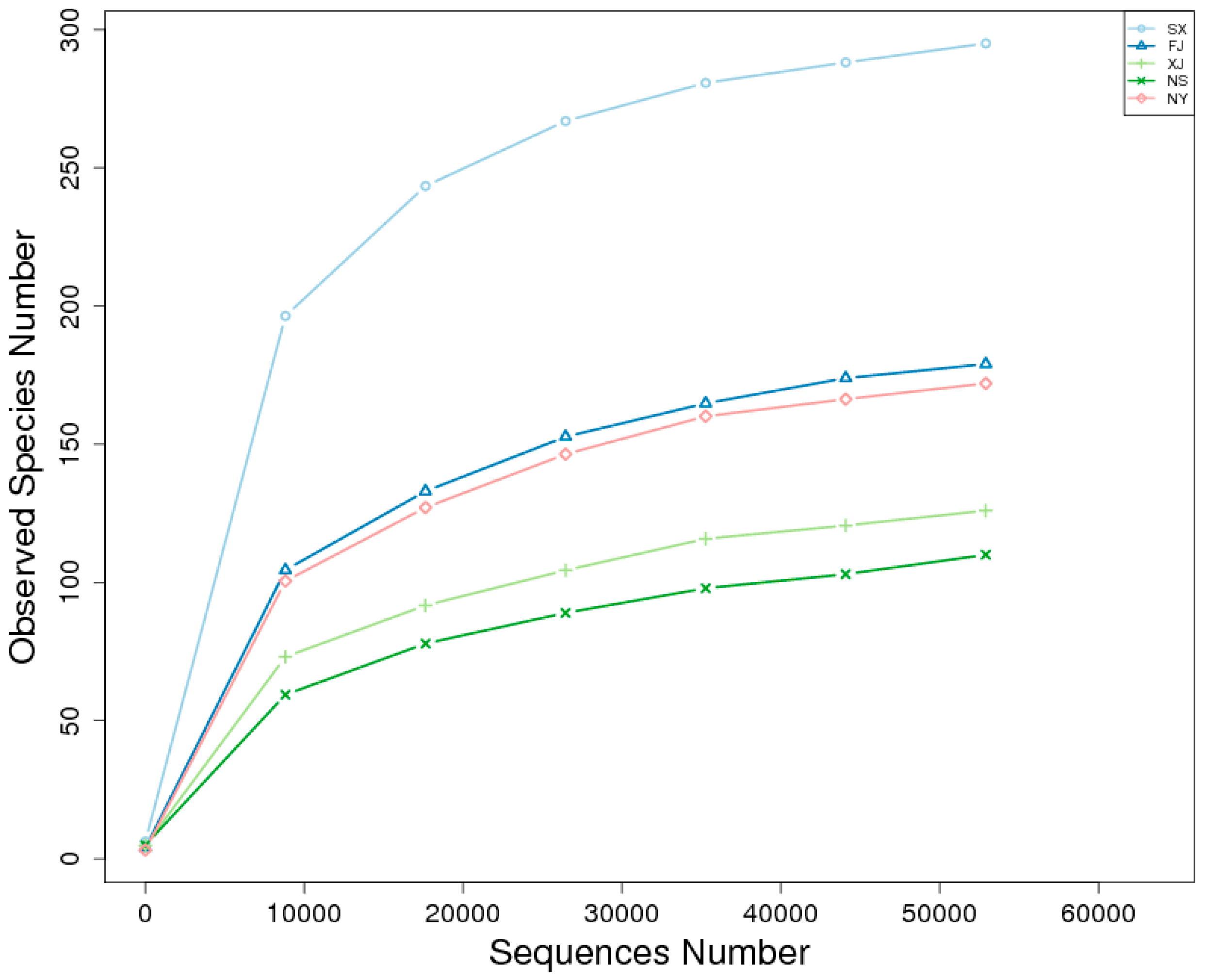
3.2. Alpha Diversity Analysis
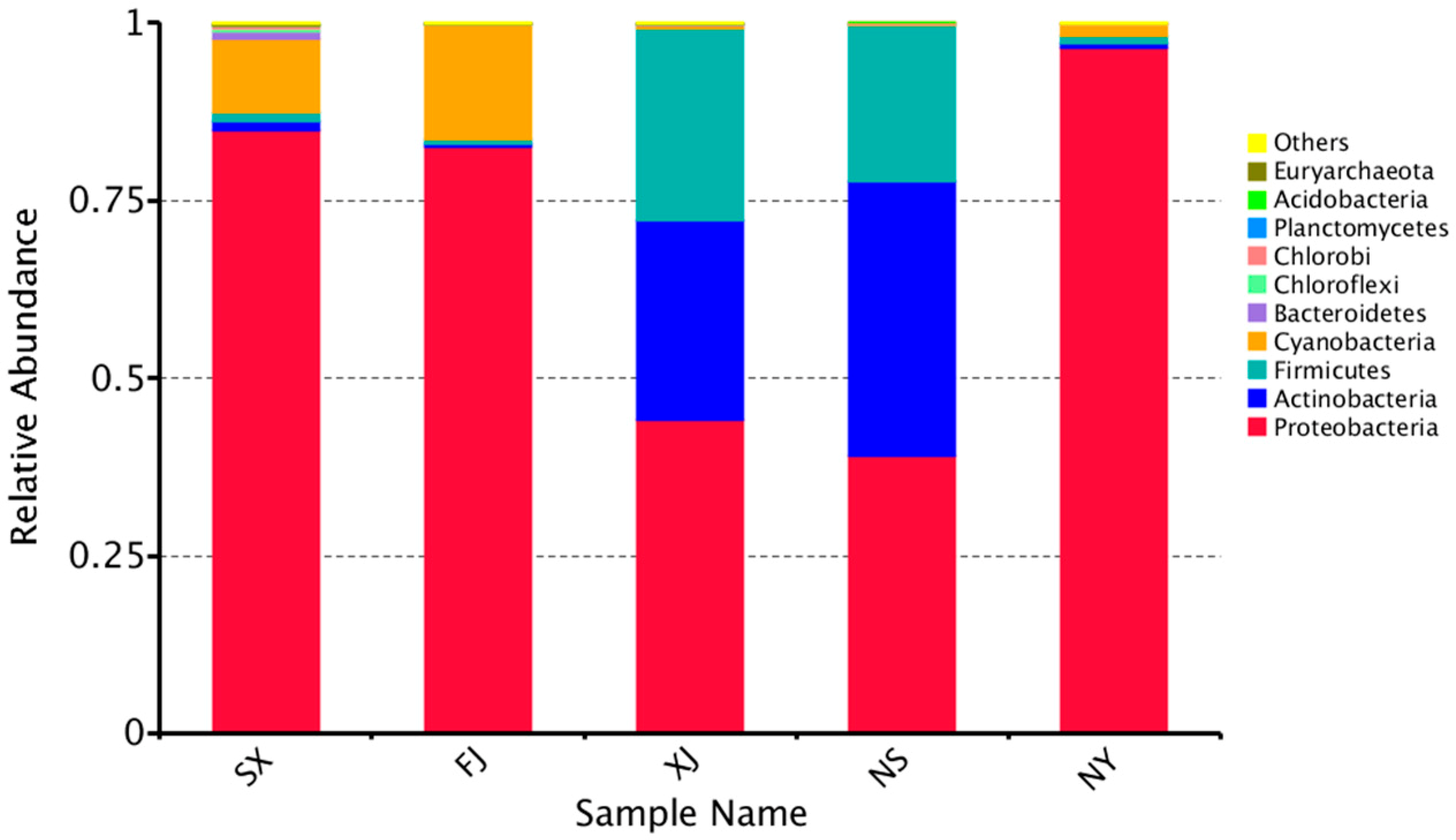
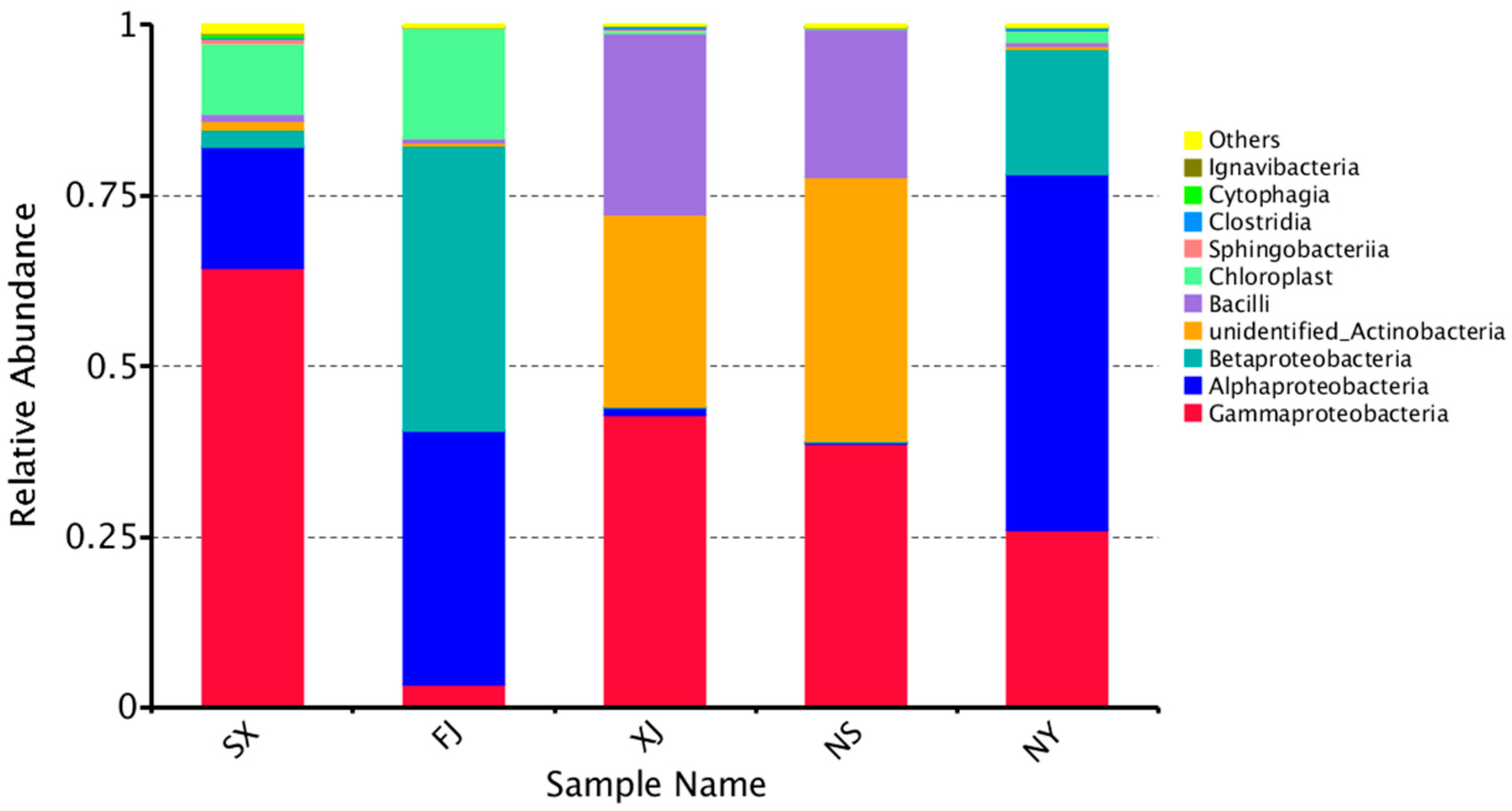
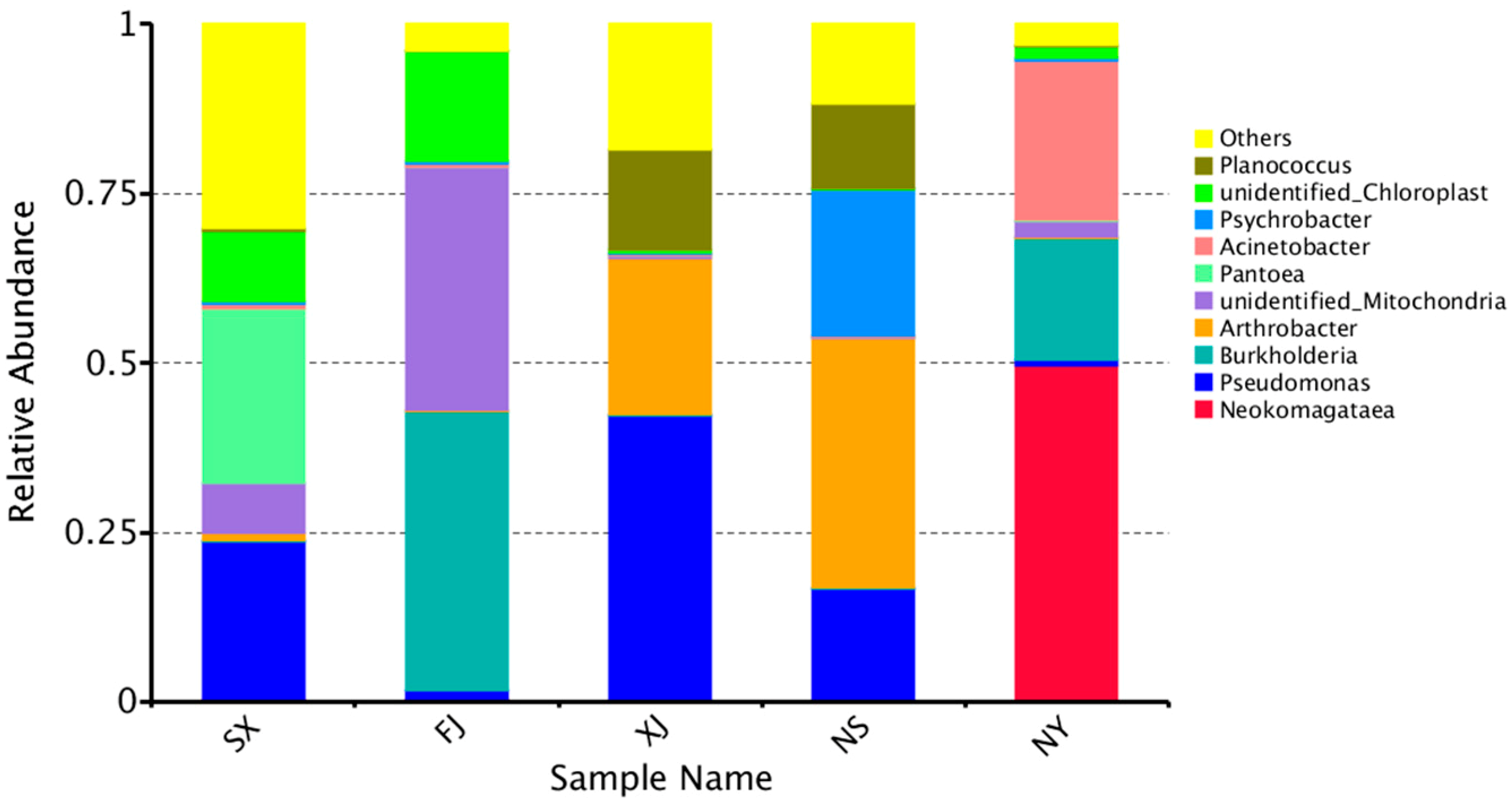
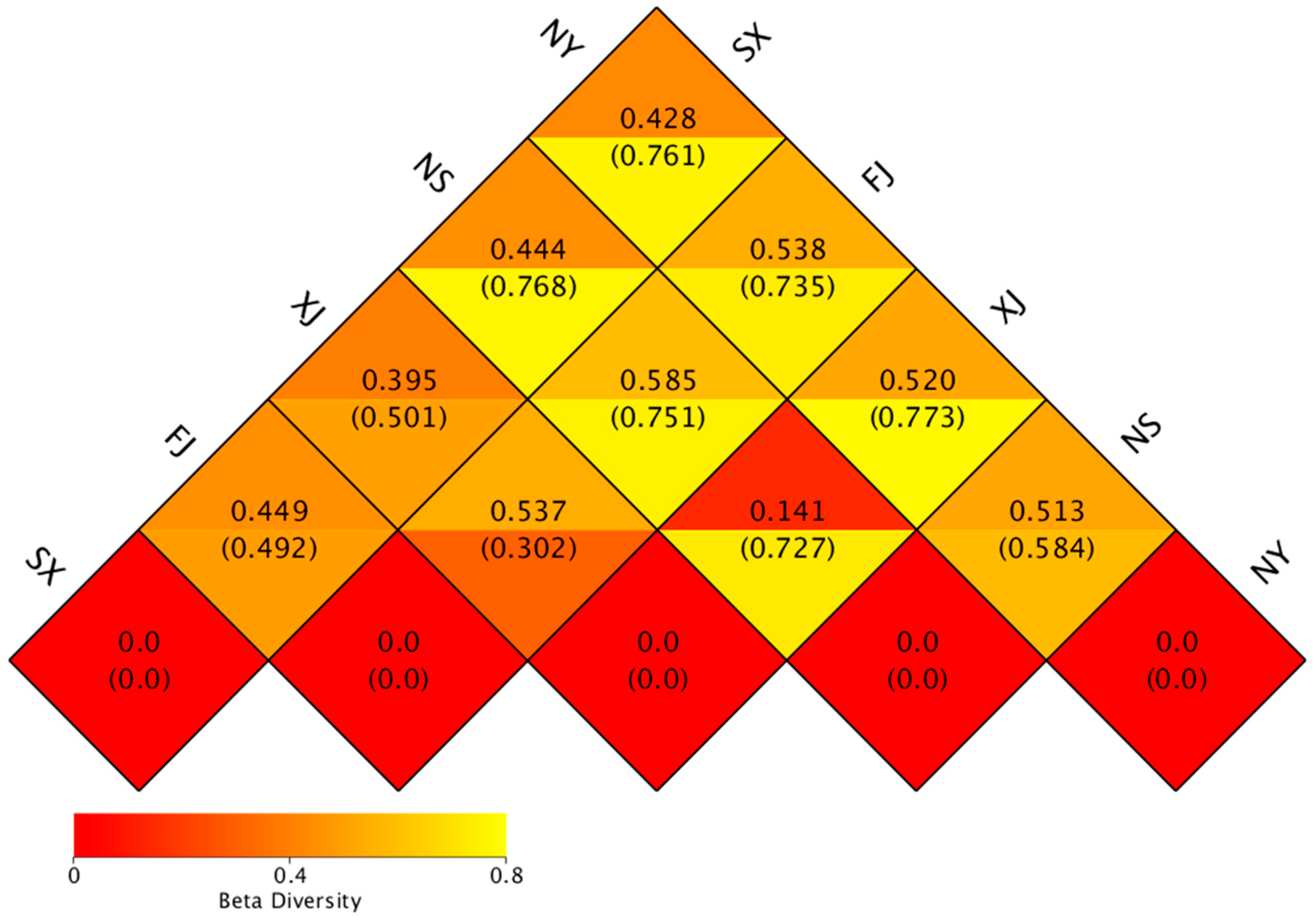
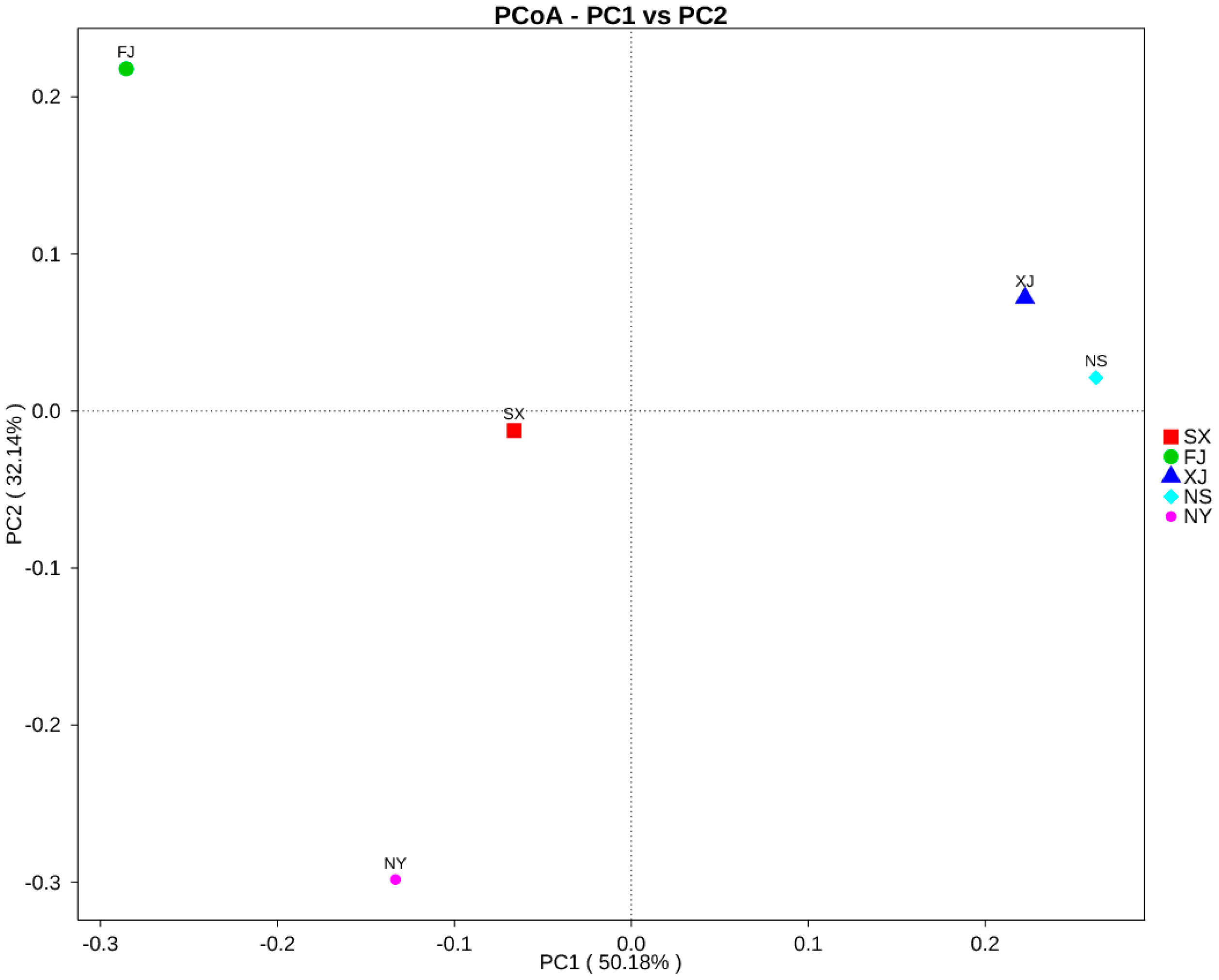
3.3. Beta Diversity Analysis
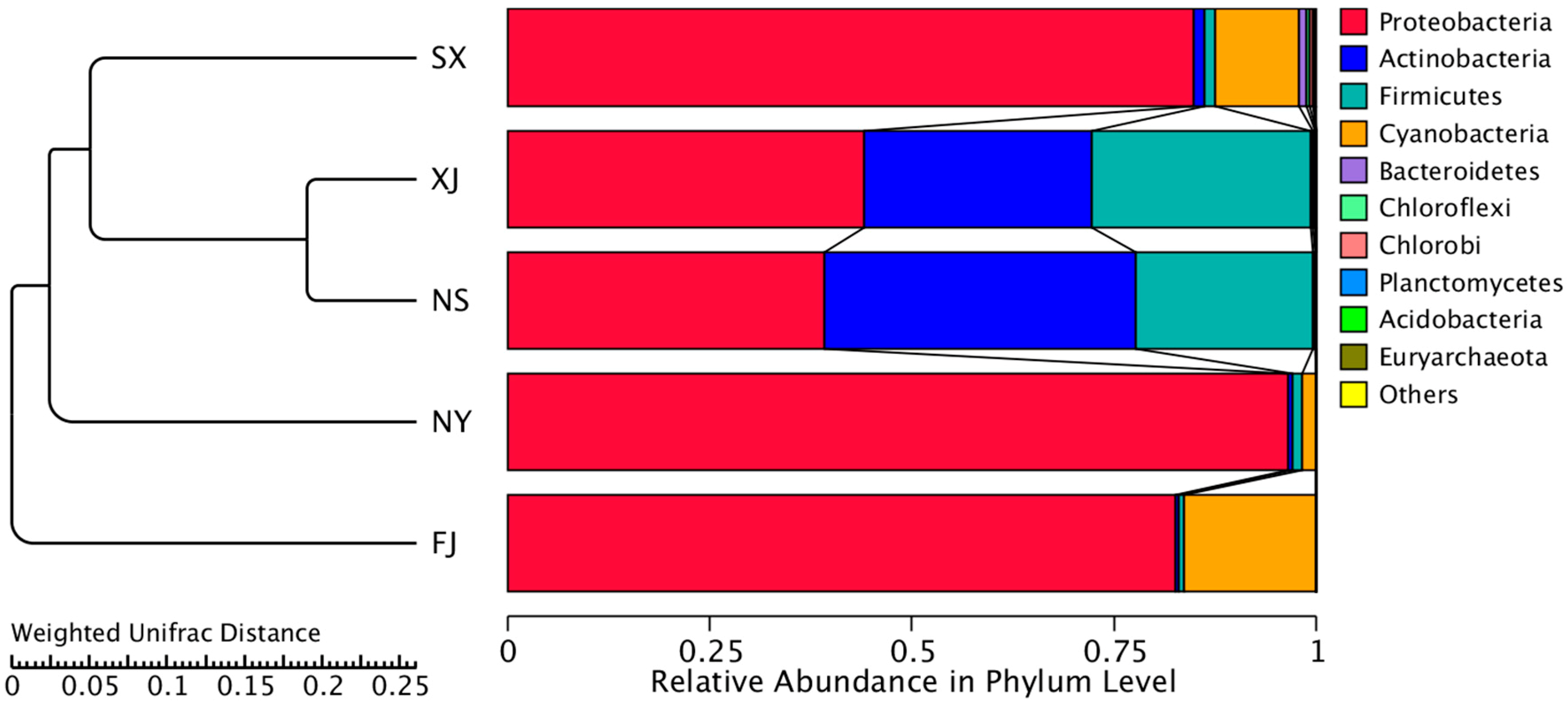
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Rosenblueth, M.; Martínez-Romero, E. Bacterial endophytes and their interaction with hosts. Mol. Plant-Microbe Interact. 2006, 19, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Mitter, B.; Colli-Mull, J.G.; Gangl, H.; Sessitsch, A. Endophytes of grapevine flowers, berries, and seeds: Identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb. Ecol. 2011, 62, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.P.; Germaine, K.; Franks, A.; Ryan, D.J.; Dowling, D.N. Bacterial endophytes: Recent developments and applications. FEMS Microbiol. Lett. 2008, 278, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Garafola, C.; Monchy, S.; Newman, L.; Hoffman, A.; Weyens, N.; Barac, T.; Vangronsveld, J.; van der Lelie, D. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of Poplar trees. Appl. Environ. Microbiol. 2009, 75, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Loaces, I.; Ferrando, L.; Scavino, A.F. Dynamics, diversity and function of endophytic siderophore-producing bacteria in rice. Microbial. Ecol. 2011, 61, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Charles, T.C.; Glick, B.R. Isolation and characterization of new plant growth- promoting bacterial endophytes. Appl. Soil Ecol. 2012, 61, 217–224. [Google Scholar] [CrossRef]
- Natul, N.; Sharma, G.D.; Barooah, M. Screening of endophytic bacterial isolates of tea (Came-llia sinensis L.) roots for their multiple plant growth promoting activities. Int. J. Agric. Environ. Biotechnol. 2013, 6, 211–215. [Google Scholar]
- Cabanás, C.G.-L.; Schilirò, E.; Valverde-Corredor, A.; Mercado-Blanco, J. The biocontrol endophytic bacterium Pseudomonas fluorescens PICF7 induces systemic defense responses in aerial tissues upon colonization of olive roots. Front. Microbiol. 2014, 5, 427. [Google Scholar]
- Lu, Q.L.; Tang, L.R.; Wang, S.; Huang, B.; Chen, Y.D.; Chen, X.R. An investigation on the characteristics of cellulose nanocrystals from pennisetum sinese. Biomass Bioenergy 2014, 70, 267–272. [Google Scholar] [CrossRef]
- Xu, Q.L.; Hunag, Z.J.; Wang, X.M.; Cui, L.H. Pennisetum sinese, roxb and pennisetum purpureum, schum.; as vertical-flow constructed wetland vegetation for removal of N and P from domestic sewage. Ecol Eng. 2015, 83, 120–124. [Google Scholar] [CrossRef]
- Hu, L.; Wang, R.; Liu, X.L.; Xu, B.; Xie, T.H.; Li, Y.Y.; Wang, M.K.; Wang, G.; Chen, Y.H. Cadmium phytoextraction potential of king grass (pennisetum sinese roxb.) and responses of rhizosphere bacterial communities to a cadmium pollution gradient. Environ. Sci. Pollut. Res. 2018, 22, 21671–21681. [Google Scholar] [CrossRef]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Giraffa, G.; Neviani, F. DNA-based, culture-independent strategies for evaluating microbial communities in food-associated ecosystems. Int. J. Food Microbiol. 2001, 67, 19–34. [Google Scholar] [CrossRef]
- Berg, G. Plant-microbe interactions promoting plant growth and health:perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Duffy, B.; Nowak, J.; Clement, C.; Barka, E.A. Use of plant growth-promoting bacteria for biocontrol of plant diseases: Principles, mechanisms of action, and future prospects. Appl. Environ. Microbiol. 2005, 71, 4951–4959. [Google Scholar] [CrossRef] [PubMed]
- USDA. Soil Survey Laboratory Methods Manual; version 3.0; Soil Survey Investigations Report No. 42; United States Department of Agriculture, Natural Resources Conservation Service, National Soil Survey Centre: Lincoln, NE, USA, 1996.
- Berry, D.; Mahfoudh, K.B.; Wagner, M.; Loy, A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl. Environ. Microbiol. 2011, 77, 7846–7849. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 1, e1. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of highthroughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011, 27, 2194. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Ear, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, P.; Hu, D.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high through-put. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M. and Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, A.; de los Dolores Soto del Rio, M.; Civera, T.; Pattono, D.; Cardazzo, B.; Bottero, M.T. Characterization of microbiota in plaisentif cheese by high-throughput sequencing. LWT-Food Sci Technol. 2016, 69, 490–496. [Google Scholar] [CrossRef]
- Zhang, L.H.; Wang, S.J. Bacterial community diversity on in-shell walnut surfaces from six representative provinces in China. Sci. Rep. 2017, 7, 10054. [Google Scholar] [CrossRef]
- Lin, B.S.; Song, Z.Z.; Zhang, L.L.; Fan, J.L.; Lin, Z.X. Composition diversity and differences of endophytic bacteria in root, stem and leaf at different growth stages of pennisetum sp. J. Fujian Agric. For. Univ. 2018, 3, 352–359. [Google Scholar]
- Redford, A.J.; Bowers, R.M.; Knight, R.; Linhart, Y.; Fierer, N. The ecology of the phyllosphere: Geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 2010, 12, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Loganathachetti, D.S.; Sadaiappan, B.; Poosakkannu, A.; Muthuraman, S. Pyrosequencing-based seasonal observation of prokaryotic diversity in pneumatophore-associated soil of avicennia marina. Curr. Microbiol. 2016, 72, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Castrillo, G.; Herrera Paredes, S.; Salas González, I.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, P.; Silva, C.; Lorite, M.J.; Izaguirre-Mayoral, M.L.; Bedmar, E.J.; Martínez-Romero, E. Molecular systematics of rhizobia based on maximum likelihood and Bayesian phylogenies inferred from rrs, atpD, recA and nifH sequences, and their use in the classfication of Sesbania microsymbionts from Venezuelan wetlands. Syst. Appl. Microbiol. 2005, 28, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Diouf, D.; Samba-Mbaye, R.; Lesueur, D.; Ba, A.T.; Dreyfus, B.; de Lajudie, P.; Neyra, M. Genetic diversity of Acacia seyal Del. Rhizobial populations indigenous to Senegalese soils in relation to salinity and pH of the sampling sites. Microb. Ecol. 2007, 54, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Moulin, L.; Munive, A.; Dreyfus, B.; Boivin-Masson, C. Nodulation of legumes by members of the beta-subclass of Proteobacteria. Nature 2001, 411, 948–950. [Google Scholar] [CrossRef]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schoning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperature grasslands and forests. Sci. RE-UPK 2016, 6, 33696. [Google Scholar] [CrossRef]
- Guo, Y.Q.; Chen, X.T.; Wu, Y.Y.; Zhang, L.; Cheng, J.M.; Wei, G.H.; Lin, Y.B. Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci. Total Environ. 2018, 1, 598–606. [Google Scholar] [CrossRef]
- Mano, H.; Tanaka, F.; Nakamura, C.; Kaga, H.; Morisaki, H. Culturable Endophytic Bacterial Flora of the Maturing Leaves and Roots of Rice Plants (Oryza sativa) Cultivated in a Paddy Field. Microbes Environ. 2007, 22, 175–185. [Google Scholar] [CrossRef]
- Botella, L.; Santamaría, O.; Diez, J.J. Fungi associated with the decline of Pinus halepensis in Spain. Fungal Divers. 2010, 1, 1–11. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | SX | FJ | XJ | NS | NY |
|---|---|---|---|---|---|
| Yanan, Shaanxi Province | Fuzhou, Fujian Province | Jichang Xinjiang Uygur Autonomous Prefecture | Bayannaoer, Inner Mongolia (sandy land) | Bayannaoer, Inner Mongolia (saline-alkali land) | |
| Altitude | 1000 m | 600 m | 560 m | 1500 m | 1500 m |
| Latitude and longitude | 36°63′97″ N, 109°32′07″ E | 26°08′55″ N, 119°28′22″ E | 44°04′21″ N, 87°18′19″ E | 40°13′55″ N, 107°05′05″ E | 40°30′19″ N, 107°02′98″ E |
| Annual average temperature (°C) | 13.0 | 19.0 | 6.0 | 4.0 | 4.0 |
| Annual average precipitation (mm) | 576.9 | 1700.0 | 150.0 | 50.0 | 50.0 |
| Total nitrogen (%) | 0.62 | 0.53 | 0.24 | 0.12 | 0.04 |
| Total phosphorus (mg/kg) | 1784 | 2065 | 2036 | 1465 | 1718 |
| Effective phosphorus (mg/kg) | 12.6 | 10.0 | 10.8 | 3.0 | 3.5 |
| Available potassium (mg/kg) | 132.9 | 112.8 | 324.7 | 154.1 | 99.5 |
| Total carbon (%) | 2.47 | 4.00 | 1.44 | 0.77 | 0.23 |
| pH | 8.35 | 6.98 | 8.27 | 8.9 | 9.38 |
| Raw Tags | Clean Tags | Effective Tags | Base (nt) | AvgLen (nt) | Q20 | GC% | Effective% |
|---|---|---|---|---|---|---|---|
| 89,003 | 67,460 | 63,306 | 26,672,103 | 421 | 98.24 | 54.11 | 71.13 |
| 92,592 | 70,360 | 69,420 | 28,879,554 | 416 | 98.28 | 55.08 | 74.97 |
| 88,774 | 69,217 | 62,597 | 26,450,228 | 423 | 98.33 | 53.25 | 70.51 |
| 81,329 | 64,091 | 53,506 | 22,509,234 | 421 | 98.35 | 53.55 | 65.79 |
| 82,770 | 65,669 | 64,215 | 26,657,088 | 415 | 98.41 | 53.72 | 77.58 |
| Sample Name | Observed Species | Shannon | Simpson | Chao1 | ACE | Goods Coverage | PD_Whole Tree |
|---|---|---|---|---|---|---|---|
| SX | 295 | 3.795 | 0.863 | 308.097 | 309.216 | 0.999 | 38.881 |
| FJ | 179 | 2.165 | 0.677 | 192.286 | 200.258 | 0.999 | 28.621 |
| XJ | 126 | 2.982 | 0.816 | 140.130 | 145.552 | 1.000 | 23.883 |
| NS | 110 | 2.780 | 0.787 | 126.240 | 145.058 | 0.999 | 10.837 |
| NY | 172 | 2.161 | 0.665 | 182.800 | 190.536 | 0.999 | 16.980 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Z.-S.; Zhang, B.-C.; Qi, X.-Y.; Sun, Z.-H.; He, X.-L.; Liu, Y.-Z.; Li, J.; Chen, K.-K.; Lin, Z.-X. Root-Associated Endophytic Bacterial Community Composition of Pennisetum sinese from Four Representative Provinces in China. Microorganisms 2019, 7, 47. https://doi.org/10.3390/microorganisms7020047
Deng Z-S, Zhang B-C, Qi X-Y, Sun Z-H, He X-L, Liu Y-Z, Li J, Chen K-K, Lin Z-X. Root-Associated Endophytic Bacterial Community Composition of Pennisetum sinese from Four Representative Provinces in China. Microorganisms. 2019; 7(2):47. https://doi.org/10.3390/microorganisms7020047
Chicago/Turabian StyleDeng, Zhen-Shan, Bao-Cheng Zhang, Xiang-Ying Qi, Zhi-Hong Sun, Xiao-Long He, Yu-Zhen Liu, Jing Li, Kai-Kai Chen, and Zhan-Xi Lin. 2019. "Root-Associated Endophytic Bacterial Community Composition of Pennisetum sinese from Four Representative Provinces in China" Microorganisms 7, no. 2: 47. https://doi.org/10.3390/microorganisms7020047
APA StyleDeng, Z. -S., Zhang, B. -C., Qi, X. -Y., Sun, Z. -H., He, X. -L., Liu, Y. -Z., Li, J., Chen, K. -K., & Lin, Z. -X. (2019). Root-Associated Endophytic Bacterial Community Composition of Pennisetum sinese from Four Representative Provinces in China. Microorganisms, 7(2), 47. https://doi.org/10.3390/microorganisms7020047